
Angle-closure glaucoma with attenuated mucopolysaccharidosis type l in a Chinese family
2021-11-30 04:48:04YanLiuLiDaiRanLongFuQuanChaoGuLingYu
Yan Liu, Li Dai, Ran Long, Fu Quan, Chao Gu, Ling Yu
1Department of Ophthalmology, the Affiliated Hospital of Southwest Medical University, Luzhou 646000, Sichuan Province, China
2Department of Radiology, the Affiliated Hospital of Southwest Medical University, Luzhou 646000, Sichuan Province, China
Dear Editor,
I am Dr. Yan Liu, from the Department of Ophthalmology,the Affiliated Hospital of Southwest Medical University,Luzhou, China. I write to present a rare case of angle-closure glaucoma (ACG) with attenuated mucopolysaccharidosis type I (MPS I).
MPS I is a metabolic disease that involved in the degradation of glycosaminoglycan (GAG). It causes a deficiency of the lysosomal enzyme a-L-iduronidase (IDUA)viaa pathogenic mutation in the IDUA gene, which is situated on chromosome 4p16.3 and contains 14 exons and encodes a 653 amino-acidlong polypeptide. This condition causes the accumulation of dermatan heparan sulfate (DS) and heparan sulfate (HS) in cells in most tissues, which leads to progressive multiorgan dysfunction[1]. The common symptoms include characteristic coarse facial features, joint stiffness, corneal clouding,hepatosplenomegaly, hearing loss, heart disease, respiratory infection, and a reduced lifespan. MPS I is now likely to be described as attenuated MPS I (mild clinical manifestations and a normal life span) or severe MPS I (severe central nervous system complications and a short life span)[2]. In accordance with the Declaration of Helsinki, informed consent was obtained from the patient before the study. This study was approved by the Institutional Review Board of the Southwest Medical University (permitted number: KY201911).
The proband was a 36-year-old Chinese male with progressive loss of binocular vision and eye pain for 5y. On initial examination, his best corrected visual acuity (BCVA) was 0.3 logMAR (OD) and 0.6 logMAR (OS), and refraction was+6.00/+1.00×70° in the right eye and +6.50/+0.5×140° in the left eye. A slit-lamp examination revealed ground-glass opacity in the corneas, a shallow peripheral anterior chamber [<1/4 corneal thickness (CT)] and transparent lens (Figure 1A and 1B).The fundus examination showed that the cup-disk ratios were 0.3 and 0.8 in the right and left eyes, respectively (Figure 1C and 1D). According to Goldmann-applanation tonometry, the intraocular pressures (IOPs) were 22 mm Hg (OD) and 45 mm Hg(OS). Optical coherent tomography (OCT) suggested thinning of the retinal nerve fiber layer (RNFL), and ultrasound biomicroscopy (UBM) showed that the anterior chamber depth measurements were 1.81 mm (OD) and 1.86 mm (OS)and the angle was narrow in both eyes (Figure 1E and 1F); a field of vision investigation of the left eye showed a tubular field of vision. The IOL-Master analysis showed that the axial lengths were 19.85 mm (OD) and 20.20 mm (OS), the central corneal thicknesses (CCTs) were 626 μm (OD) and 632 μm(OS), and the white-to-white distances (WTWs) were 11.4 mm(OD) and 11.6 mm (OS).
The following phenotypic features were noted: coarse facial features, a short neck, deformity of the spine and fingers,short stature, stiff joints, claw-shaped hands, and normal intelligence (Figure 1G and 1H). Considering the proband’s clinical history and auxiliary examination, a diagnosis of attenuated MPS I with ACG was suspected. An X-ray examination showed the following: broad and short bilateral clavicular scapulas, secondary bilateral shoulder joint degeneration, a straightened thoracic curvature and broadened bilateral ribs. An electrocardiogram revealed left bias in the pseudoelectric axis. Echocardiography suggested cardiac valvular disease, moderate mitral and aortic stenosis with mild regurgitation, mild reflux of the tricuspid valve, and left ventricular diastolic dysfunction. An abdominal ultrasound examination showed hepatomegaly and a normal spleen. A urine mucopolysaccharide qualitative test was positive.
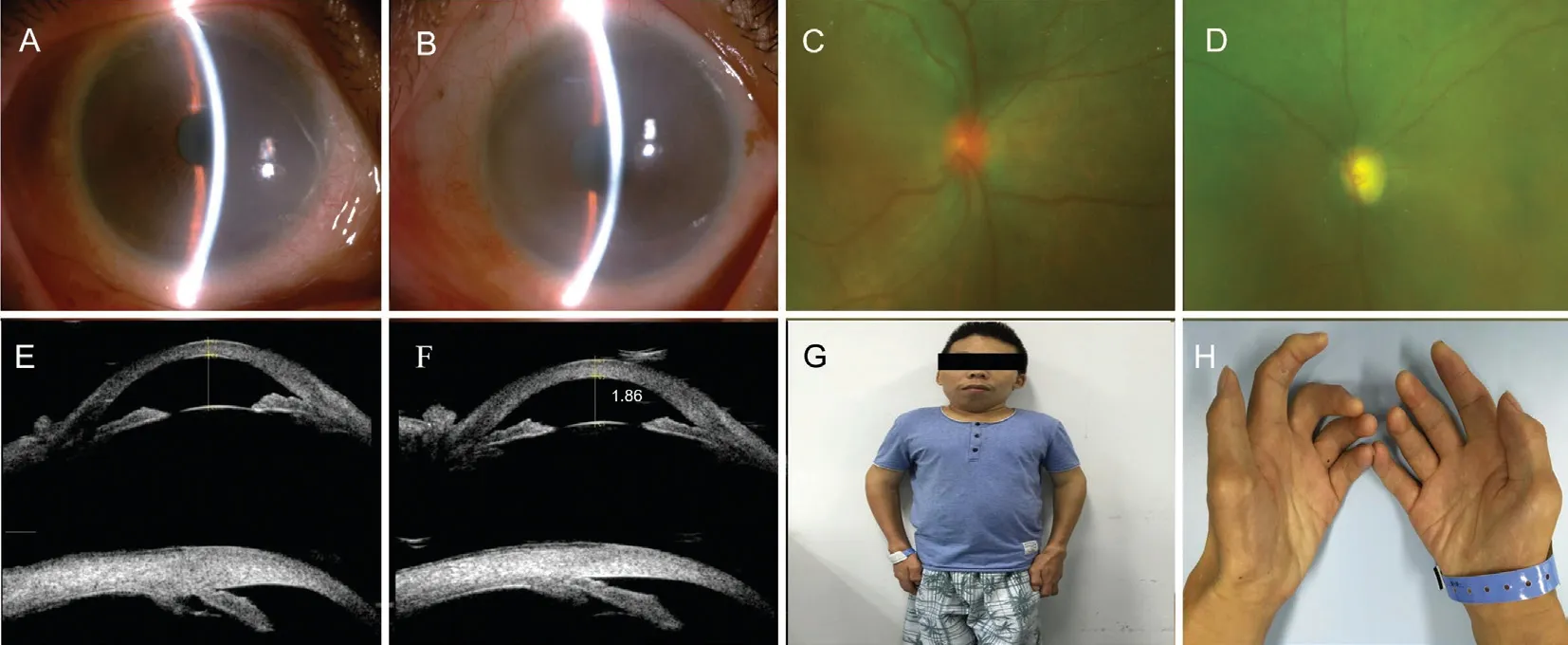
Figure 1 The patient complained of progressive loss of binocular vision and eye pain for 5y Slit-lamp photographs of the anterior segment of the right eye (A) and the left eye (B) were demonstrating cloudy corneas and the shallow anterior chamber. Scanning laser ophthalmoscopey showed the condition of the fundus, which the cup-disc ratio was 0.3 in the right eye (C) and 0.8 in the left eye (D). Ultrasound biomicroscopy(UBM) demonstrating the anterior chamber were shallow and the angle were narrow in both eyes (E-F). The patient was short-statured, had coarse facial features and claw-shaped hands (G-H).
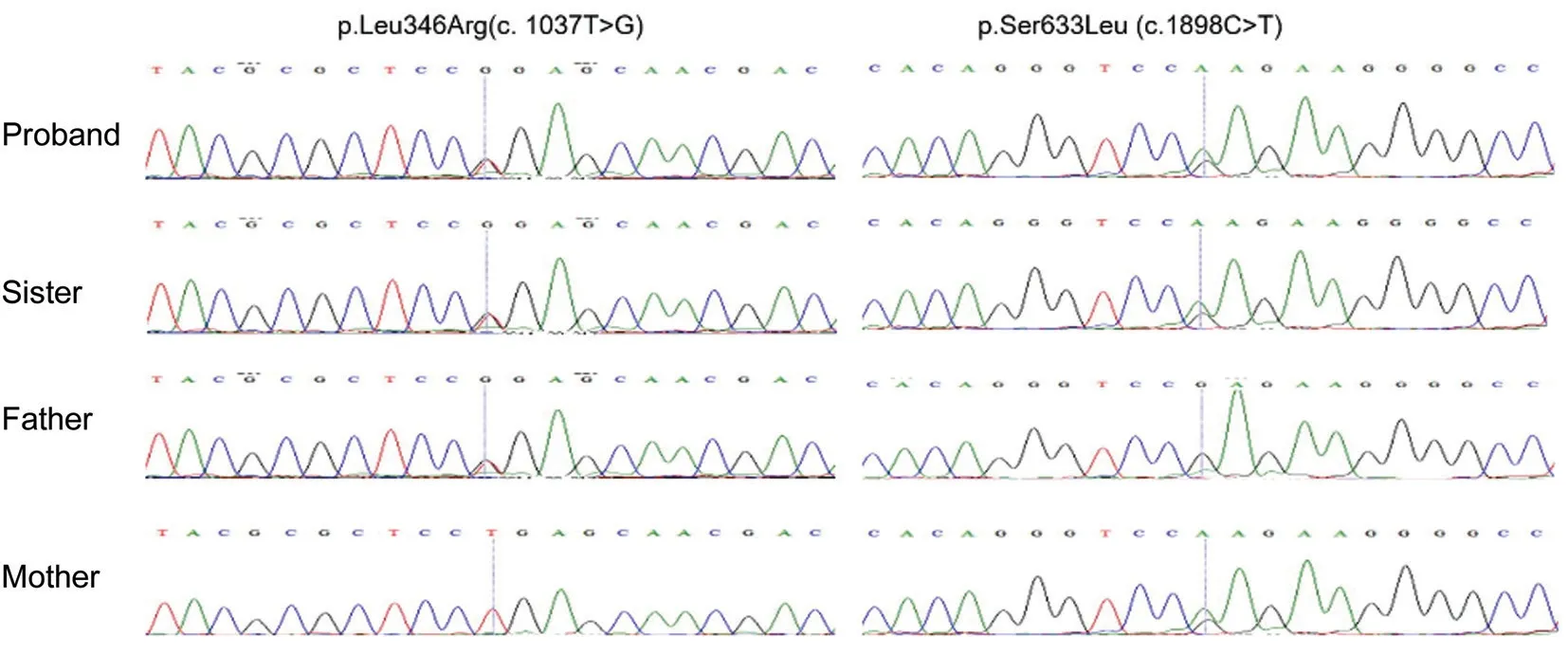
Figure 2 The IDUA mutation p.Leu346Arg (c.1037T>G) in the proband and his sister and father as indicated, normal sequencing maps for his mother. The IDUA mutation p.Ser633Leu (c.1898C>T) in the proband and his sister and mother as indicated, normal sequencing maps for his father.
The proband’s sister, who recently died of heart failure, had similar clinical manifestations: short stature, facial coarsening,substantial skeletal and joint deformities, severe heart attacks.The proband’s parents were healthy and denied a consanguineous marriage. No abnormalities were found in their eyes.
Genomic DNAs (gDNAs) were isolated from the 2 mL peripheral blood samples taken from the proband, his sister and his parents by using QIAamp DNA Blood kits(Qiagen, Hilden, Germany). All the procedures followed the manufacturer’s standard. The gDNA was sequenced by illumina hiseq 4000. The data analysis followed DOI: 10.1126/science.aao6575. The Figure 2 was the result of SNV filter,and then, all the family members’ gDNAs were detected by Sanger sequencing. Gene sequencing showed that the proband and his sister (both patients) carried heterozygous mutations in the IDUA gene that caused mucopolysaccharidosis (both NM_000203: exon8:c.1037T>G:p.L346R and NM_000203:exon14:c.1898C>T:p.S633L). His father carried only the heterozygous mutation NM_000203: exon8:c.1037T>G:p.L346R, and his mother carried only the heterozygous mutation NM_000203: exon14:c.1898C>T:p.S633L, consistent with the rule of compound heterozygous inheritance.
The patient was treated with intravenous 20% mannitol,topical anti-glaucoma drops and pilocarpine eye drops were administered, IOP dropped to 20 mm Hg in both eyes. The trabeculectomy was subsequently performed in both eyes.On the 7thpostoperative day, BCVA were not improved in both eyes, IOP decreased to 18 mm Hg (OD) and 20 mm Hg (OS) and the patient’s eye pain was relieved. One year postoperatively, his BCVA was 0.4 logMAR (OD) and 0.8 logMAR (OS), the corneal clouding was progressed, the visual field progressed, and the IOP values were 15 mm Hg(OD) and 18 mm Hg (OS), with three glaucoma medications used in both eyes.
The signs and symptoms of MPS I, which is the representative prototype of MPS, are the most typical. Ocular involvement is one of the early manifestations of MPS, and ophthalmologists may be the first to diagnose MPS[3]. Ultrastructure and histochemistry techniques have shown that GAG deposits may occur in the conjunctiva, cornea, sclera, lens epithelial cells,choroid, intracellular and extracellular matrix of the ciliary body nonpigmented epithelial cells,etc., resulting in various clinical signs in eyes.
Studies have shown that glaucoma is an early complication of MPS. The prevalence of glaucoma in the MPS I population is estimated to be 10%[4]. Quigleyet al[5]described two siblings with MPS as well as acute ACG and found that corneal limbus specimens showed excessive adenosine monophosphate(AMP) deposition. Abnormal thickening of the anterior segment structure may result in physical obstruction of the angle. Moreover, the abnormal deposition of AMP in the trabecular meshwork may be the pathogenesis of glaucoma.Spellacyet al[6]reported a case of MPS I with an IOP of 41 mm Hg. Ultrastructural analysis of the tissues and iris at the corneoscleral junction revealed obstruction of aqueous humor outflow, which was caused by cell swelling in the trabecular meshwork near Schlemm’s canal and the formation of abnormal vesicles. Nowaczyket al[7]concluded that glaucoma may be an early complication of Hurler’s syndrome based on cases of glaucoma in infancy. It was found that the accumulation of GAG caused hyperemia and deformation of the trabecular meshwork, causing thickening of the sclera and corneal extracellular matrix, as well as changes in the corneoscleral limbus and adjacent Schlemm’s canal interfering with aqueous humor outflow. Previous studies revealed that ACG can be caused by angle narrowing in MPS due to GAG accumulation in anterior chamber tissues such as the cornea,iris, and ciliary body, while open angle glaucoma (OAG)can be induced by the obstruction of aqueous humor outflow induced by GAG accumulation in trabecular meshwork cells[3-6]. In addition, the formation of cysts in different parts of the iris and ciliary body (membrane-bound vacuoles in the nonpigmented epithelium of the ciliary body) was also considered to be one of the causes of glaucoma[8].
Hematopoietic stem cell transplantation (HSCT) is recommended therapy for severe MPS I patients before 2y of age, which could protect the nervous system and prolong life. Enzyme replacement therapy (ERT) with laronidase (recombinant human IDUA) improves disease manifestations (e.g.,pulmonary function, joint mobility, cardiac disease), which is effective across a wide range of ages[9-10]. HSCT and ERT can extend a patient’s life, ameliorate symptoms, and improve the quality of life, which indicate the great importance of the precise early diagnosis of MPS. Although eye lesions are an early complication of MPS, the complexity of the disease makes it difficult to assess and manage, which delays the diagnosis and treatment of eye lesions. Therefore, it is of great value to enhance ophthalmologists’ understanding of MPS and establish a systematic guideline for ophthalmological diagnosis and treatment for patients with MPS.
In addition, genetic counseling should be provided as MPS I in an autosomal recessive manner. Molecular genetic testing could identify IDUA disease-causing variant, which to clarify genetic status for family members. Carrier testing and prenatal testing for at-risk family members, who carry diseasecausing IDUA variants, could predict risk rate and propose the countermeasure[11].
ACKNOWLEDGEMENTS
Foundations:Supported by the National Natural Science Foundation of China (No.81570841); the Southwest Medical University Affiliated Hospital Foundation (No.16004).
Conflicts of Interest:Liu Y,None;Dai L,None;Long R,None;Quan F,None;Gu C,None;Yu L,None.
 International Journal of Ophthalmology2021年11期
International Journal of Ophthalmology2021年11期
- International Journal of Ophthalmology的其它文章
- Toric implantable collamer lens for the management of pseudophakic anisometropia and astigmatism
- Novel biallelic compound heterozygous mutations in FDXR cause optic atrophy in a young female patient: a case report
- A novel temporary keratoprosthesis technique for vitreoretinal surgery
- Human umbilical cord-derived mesenchymal stem cells treatment for refractory uveitis: a case series
- lntroduction of longstanding complicated sulcus intraocular lens into the intact capsular bag
- Applications of dynamic visual acuity test in clinical ophthalmology