
Novel biallelic compound heterozygous mutations in FDXR cause optic atrophy in a young female patient: a case report
2021-11-30 04:48:02SiJiaSongYingHongKeXuChunZhang
Si-Jia Song, Ying Hong, Ke Xu, Chun Zhang
1Department of Ophthalmology, Peking University Third Hospital, Beijing 100191, China
2Beijing Key Laboratory of Restoration of Damaged Ocular Nerve, Peking University Third Hospital, Beijing 100191,China
Dear Editor,
I write to present a case of novel biallelic compound heterozygous mutations in FDXR cause optic atrophy in a young female patient. This is a rare genetic mutation with new site mutation.
Auditory neuropathy and optic atrophy (ANOA, OMIM:617717) is an autosomal recessive disease that is caused by ferredoxin reductase (FDXR, OMIM:103270) gene mutation. This rare disease was first reported in 2017 by Paulet al[1], who examined 2 boys and 6 girls presenting ANOA and conducted whole-exome sequencing, eventually revealing biallelic mutations in the FDXR gene. The shared characteristic of the 8 patients was the onset of visual and hearing impairment in childhood, in some cases complicated by very poor speech understanding. At the same time, another group reported 17 patients from 13 unrelated families; adding more clinical features of ANOA, such as ataxia, hypotonia,global developmental delay, spasticity, seizures, movement disorder, and abnormalities in magnetic resonance imaging(MRI)[2]. FDXR is a mitochondrial flavoprotein that receives electrons from NADPH, thus initiating the electron-transport chain reaction[1,3]. It is the sole human enzyme responsible for synthesizing iron-sulfur clusters and heme, which maintains normal mitochondrial function and steady energy supplementation[4]. The FDXR gene is located at 17q25 and contains 12 exons[5]. As neurons require large amounts of energy to maintain normal functions, mitochondrial dysfunction often leads to multiorgan disorders, similar to the manifestations of patients in earlier studies. We describe a patient with a biallelic compound FDXR mutation who presented optic atrophy without any other abnormalities,which may be due to novel site mutations in the FDXR gene.This patient is the only individual thus far manifesting singleorgan disorder in ANOA, and the findings constitute essential supplemental knowledge for this disease.
A 25-year-old Chinese woman who experienced blurred vision lasting 13y in both eyes visited our department. Thirteen years prior, her visual acuity in both eyes decreased gradually over 6mo without any known causes, but her visual acuity was stable thereafter. She was diagnosed with optic nerve atrophy at that time and did not receive any effective treatment. In addition, she had no family history or other diseases, especially no history of trauma or poisoning. Her visual acuity was 20/166 OU, and BCVA was 20/133 OU. Refraction was-1.50 diopter sphere (DS)/-0.75 diopter cylinder (DC)×153° in the right eye (OD) and -1.75DS/-0.50DC×55° in the left eye(OS). The color vision test for distinguishing red and green was normal. Her intraocular pressure was 13 mm Hg OD and 14 mm Hg OS. The cornea and lens were transparent, and the depth of the anterior chamber was deep. The diameter of the pupil and light reflex were normal. Fundus images showed pale optic discs, with a cup-to-disc ratio increased to 0.7(Figure 1). Optic coherence tomography (OCT) revealed that bilateral retinal nerve fibre layer (RNFL) thickness and limiting membrane-retinal pigment epithelium (ILM-RPE) thickness were diminished, but the morphology of the macular lutea was normal. The Humphrey visual field test showed temporal visual field defects in both eyes and centric scotoma in the right eye (Figure 2). According to the pattern visual evoked potential(P-VEP), P100 amplification was decreased in both eyes; flash visual evoked potential (F-VEP) displayed P2 latency that was extended in both eyes. Multifocal electroretinogram revealed that P1 amplification was lower in the left eye than in the right eye.
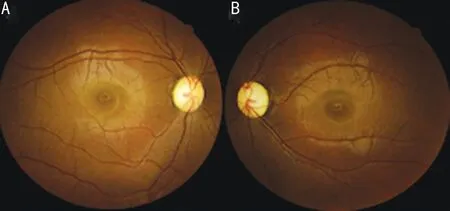
Figure 1 Fundus images A: The right eye; B: The left eye.
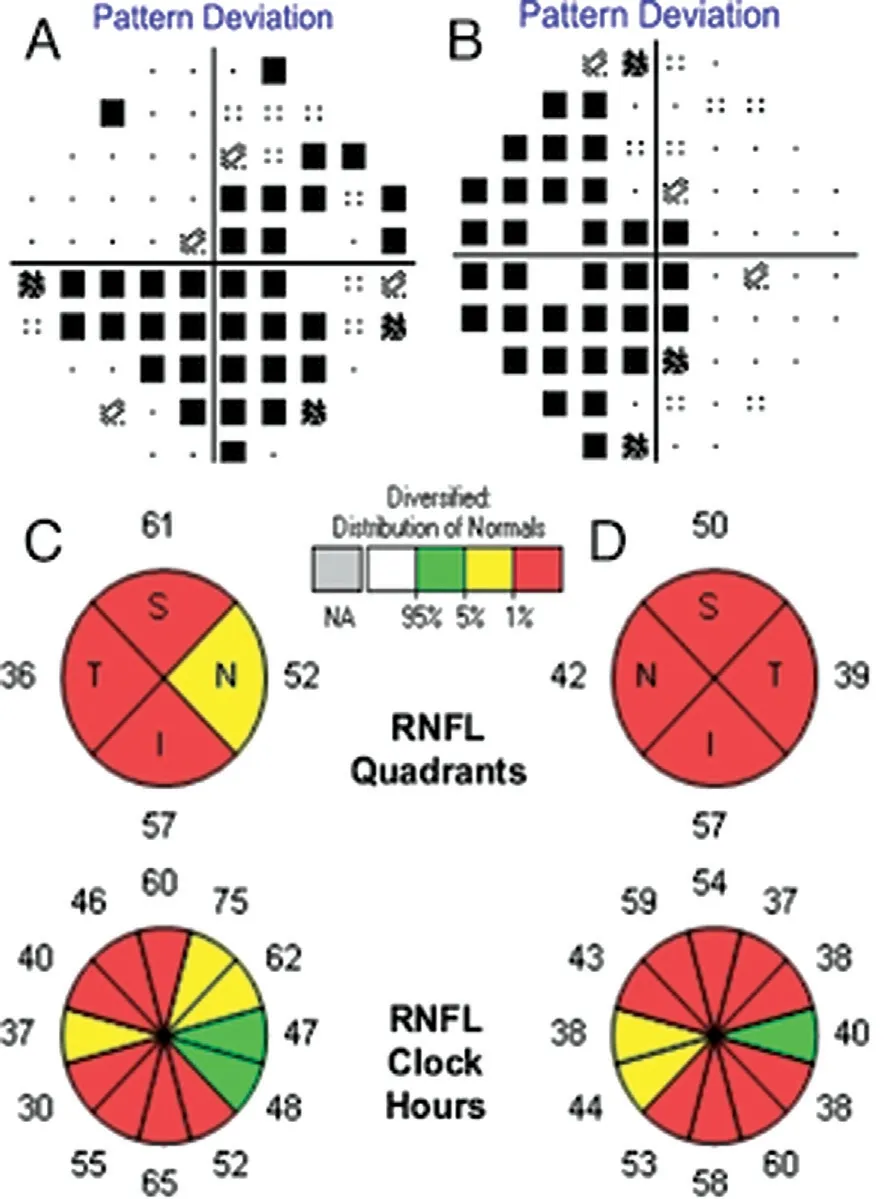
Figure 2 Humphrey visual field test and optic coherence tomography A: VF of the right eye; B: VF of the left eye; C: OCT of the right eye; D: OCT of the left eye; RNFL: Retinal nerve fiber layer;S: Superior; T: Temporal; I: Inferior; N: Nasal. The fixation loss, false positive rate and false negative rate are 2/17, 6%, 9% for the right eye and 1/15, 10%, 20% for the left eye.
Because the Humphrey visual field test showed temporal visual field defects in both eyes, we speculated it might be pituitary disorder. Neurosurgery consultation were held to carry out general examinations. Physical examination showed that there were no other cranial neurological deficits. For instance, hearing, facial sensations, and motor function of the tongue and facial muscles were all normal and free movement of the four limbs and normal muscle force and muscle strength.Based on brain MRI, the superior margin of her hypophysis was distended, but it didn’t press on optic nerve. And the height of her hypophysis was 7 mm, which was basically normal (Figure 3). Hypophysial hormone examination revealed a high level of thyroid-stimulating hormone (TSH), though free triiodothyronine (FT3) and free thyroxine (FT4) were within the normal ranges of 9.5 μIU/mL, 3.15 pg/mL and 1.12 ng/dL,respectively. Finally, pituitary disorder was excluded.
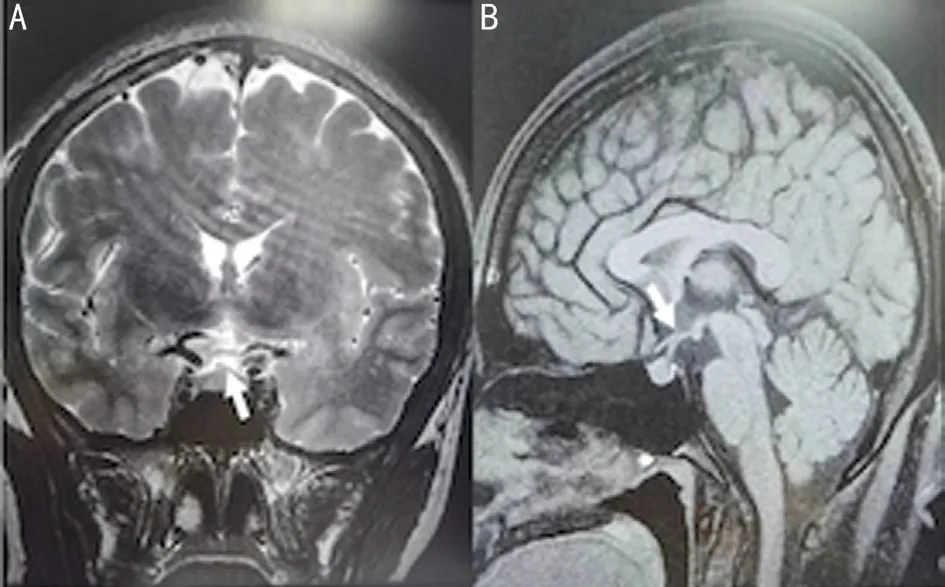
Figure 3 Brain MRI A: Coronal images; B: Sagittal images.
In view of the above examinations, we suspected it might be hereditary optic neuropathy. With the permission of the patient and her family members, we obtained fundus imaging of her parents and blood samples from her parents and sister. Visual acuity of her parents was 20/20 OU. Fundus imaging of her parents showed an increasing cup-to-disc ratio in both eyes of her father and left eye of her mother (Figure 4). Whole-exome sequencing (WES) was performed for the patient and her family members, and FDXR mutations that might be related to the disease but with unknown pathogenicity were found. Other genetic mutations included autosomal recessive heterozygous mutations and genetic variants not clearly associated with the patient’s clinical manifestations. The FDXR gene, which is on chromosome 17q25, spans 12 kb and contains 12 exons.
The exons are divided into three main domains: two NAD(P)-binding domains and a FAD/NAD(P)-binding domain in the middle. FDXR compound heterozygous mutations in exon 10 in her father and in exon 9 in her mother were confirmed, both of which are missense mutations: c.1102G>A (p.D368N) and c.940G>T (p.V314L), respectively (Figure 5). Both mutations are novel and of uncertain pathogenicity. No variation at either site was detected in her sister.
The pathogenic mechanisms of FDXR gene mutation may be energy deficiency and harmful substance accumulation.Fibroblasts in patients exhibit low oxygen consumption rates,complex activities, reduced ATP production and increased oxygen species levels[2]. In addition, evidence of inflammation has been observed in brain autopsy[6]. Further research has revealed that significant optic transport defects might be the pathogenetic basis of the observed optic atrophy[7].
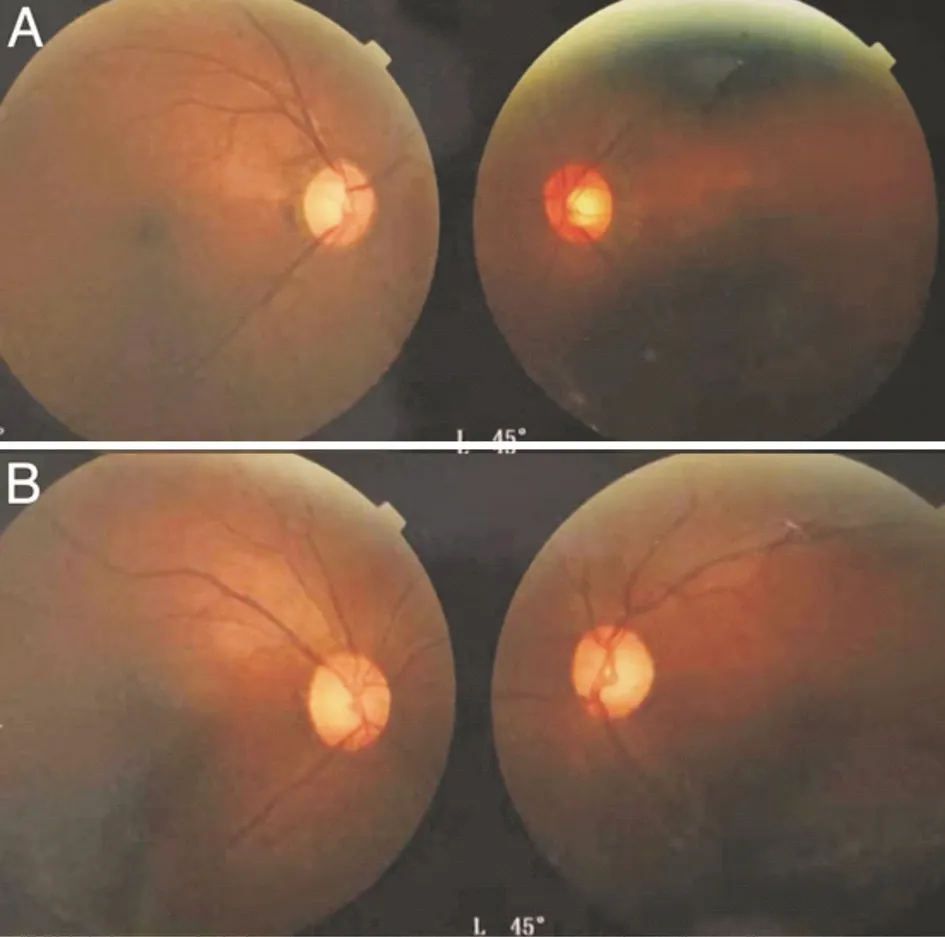
Figure 4 Fundus images of family members A: The fundus images of her father; B: The fundus images of her mother.
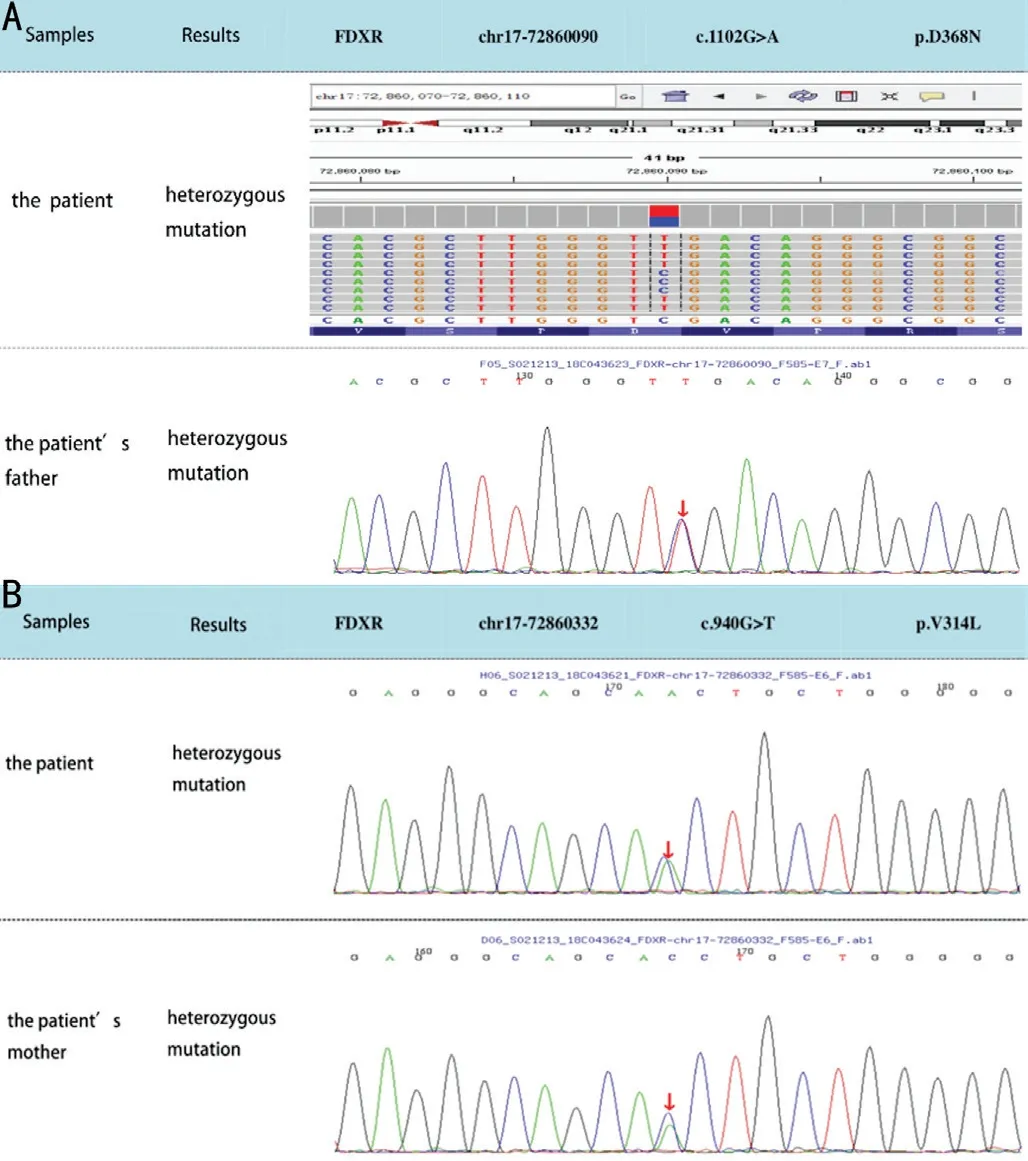
Figure 5 Results of WES A: The comparison of the patient’s mutation site and her father; B: The comparison of the patient’s mutation site and her mother.
There are several advantages for our case report. This case involves the only patient manifesting single-organ disorder in ANOA, and these findings are key for understanding the disease. Although there was no effective therapy for this patient, her correct diagnosis is extremely important for the next generation. Both biallelic FDXR genes of this patient harbored autosomal recessive mutations; thus, if her husband carries the disease gene, the risk of their children manifesting symptoms is 50%. Moreover, their children might present complication with systemic dysfunction. Therefore, assisted reproductive technology is recommended. As the FDXR gene of her sister was normal, the children of her sister did not carry the mutant gene.
There are some limitations to our case report. First, causes of optic atrophy include hereditary neuropathy, optic neuritis,glaucoma, traumatic neuropathy, and poisoning, among others[8-10]. Many other causes might have been excluded based on her history and manifestations, but it is difficult to exclude all related causes because the disease manifested 13y prior. For instance, a history of antibiotics, chemical poisons or infectious disease might be forgotten. Second, it cannot be ruled out that systemic disorders may appear in the future, thus follow-up is recommended.
ACKNOWLEDGEMENTS
Foundations:Supported by National Natural Science Foundation of China (No.81970798; No.81670851).
Conflicts of Interest: Song SJ,None;Hong Y,None;Xu K,None;Zhang C,None.
 International Journal of Ophthalmology2021年11期
International Journal of Ophthalmology2021年11期
- International Journal of Ophthalmology的其它文章
- Toric implantable collamer lens for the management of pseudophakic anisometropia and astigmatism
- Angle-closure glaucoma with attenuated mucopolysaccharidosis type l in a Chinese family
- A novel temporary keratoprosthesis technique for vitreoretinal surgery
- Human umbilical cord-derived mesenchymal stem cells treatment for refractory uveitis: a case series
- lntroduction of longstanding complicated sulcus intraocular lens into the intact capsular bag
- Applications of dynamic visual acuity test in clinical ophthalmology