
Wilson disease associated with immune thrombocytopenia:A case report and review of the literature
2019-03-14 04:17:26TianJiaoMaGuiLianSunFangYaoZhiLiangYang
World Journal of Clinical Cases 2019年17期
Tian-Jiao Ma,Gui-Lian Sun,Fang Yao,Zhi-Liang Yang
Abstract
Key words: Wilson disease;Immune thrombocytopenia;ATP7B;Case report
INTRODUCTION
Wilson Disease (WD),first described in 1912,is a hereditary genetic disorder induced by the dysfunction of copper metabolism in the liver.WD is fatal if untreated,but the prognosis can be good with timely and lifelong management[1-2].The initial signs for WD are quite varied,and some rare presentations may lead to delays in diagnosis and treatment[3-5].It is caused by mutations inATP7B,which encodes a membrane-bound P1B-type ATPase involved in copper excretion from hepatocytes.The accumulation of copper in the body can lead to multiple organ damage.ATP7Bwas first identified as the responsible gene in 1993,and now over 500 mutations have been detected and most patients with WD are compound heterozygotes[6-8].Some mutations inATP7Bshow relatively higher frequencies in special populations,such as the mutation resulting in p.Arg778Leu in the Far East[9-15].The identification of a mutation supports the diagnosis of WD,while a compound heterozygous status confirms the diagnosis.Recently,Atox1andCOMMD1were also concerned in WD patients,but there was no evidence to show their contribution[16].
Coombs-negative hemolytic anemia is the hematological presentation reported for WD and is rare.Immune thrombocytopenia (ITP) is an acquired hemorrhagic disease caused by the accelerated clearance of platelets induced by antiplatelet autoantibodies such as antiglycoprotein (GP) IIb/IIIa[17-19].WD has been associated with ITP in an adult[20],but such association has not been reported in children.
Here,we report a case of genetically-confirmed WD caused by a compoundATP7Bmutation that was inherited from the proband’s unaffected parents.The patient was diagnosed with ITP and revealed WD soon after the diagnosis of ITP,and we also discuss the association of WD with ITP.
CASE PRESENTATION
Chief complaints
The proband (Figure 1A) was an 11-year-old Chinese girl who was admitted to our hospital with chief complaints of thrombocytopenia for 15 d,and tremor in her right hand for 3 d.
History of present illness
About 15 d previously,she experienced coughing,drooling of saliva,and dysarthria without fever and was admitted to a local hospital with a diagnosis of epiglottitis.A routine blood test found a platelet count of 54 × 109/L,after which she visited a hematologist.A bone marrow examination and testing for antiplatelet antibodies were performed.HLA Class I antibody was negative,but antibodies against GP IIb/IIIa,GP Ib/Ix,and GP Ia/type IIa were positive.In bone marrow smears,granulocytic and erythrocytic series were normal,while megakaryocytes appeared immature and platelet-producing megakaryocytes were not found.According to these results,ITP was diagnosed.The child was not treated for ITP because her platelet count was greater than 50 × 109/L and she had no signs of hemorrhagic tendency.About 3 days previously,she exhibited an involuntary tremor,with numbness in her right hand.
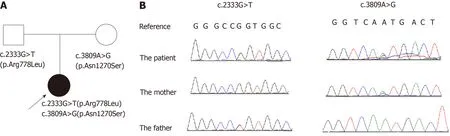
Figure1 Wilson disease patient’s family pedigree showing the mutations detected in ATP7B.
History of past illness
She was healthy in the past.
Personal and family history
Her medical and family histories were unremarkable,and she had not started menstruating.Her parents had no history of consanguinity.
Physical examination upon admission
On physical examination,she had dysarthria with normal orientation.She had no rashes,hemorrhagic signs,hepatomegaly,or splenomegaly.The pharyngeal reflex,abdominal reflexes,patellar tendon reflexes,finger-to-nose test,and Romberg’s sign were normal.Barbinski signs were negative.Alternating movements with hands were slow,and her right hand trembled.Sensitivity to heat or pain stimulation was normal.Kayser Fleischer's (K-F) rings were found under a slit lamp.
Laboratory examinations
Laboratory investigations revealed serum platelet counts of 34-43 × 109/L (normal range,100-300 × 109/L),and hemoglobin of 112-123 g/L (normal range,120-140 g/L).Liver function tests revealed alanine aminotransferase (ALT) of 59 U/L (normal range,7-40 U/L),aspartate aminotransferase (AST) of 65 U/L (normal range,13-35 U/L),albumin (ALB) of 26 g/L (normal range,40-55 g/L),total serum bilirubin of 45μmol/L (normal range,3.4-20.5 μmol/L),serum direct bilirubin (DBL) of 12.9 μmol/L(normal range,0.3-8.5 μmol/L),and plasma ammonia of 35 μmol/L (normal range,11-25 μmol/L).Her blood clotting profile showed a prothrombin time (PT) of 18.2 s(normal range,11.0-14.3 s),prothrombin time activity (PTA) of 54% (normal range,80-120 s),prothrombin time [international normalized ratio (INR)] of 1.53 (normal range,08-1.15),activated partial thromboplastin time (APTT) of 55.5 s (normal range,32.6-43.0 s),and fibrinogen of 1.27 g/L (normal range,2.0-4.0 g/l).Plasma ceruloplasmin was 190 mmol/L (normal range,220-330 mmol/L) and 24-h urine copper was 203μg/24 h (normal value,<100 μg/24 h).Her lactic acid level and urinalysis were normal.Antibodies for viral hepatitis series (including hepatitis A,B,C,D,and E viruses) were negative.Tests of immunoglobin M for Coxsackie virus,herpes virus,cytomegalovirus,Toxoplasmosis gondii,rubella virus,adenovirus,respiratory syncytial virus,andMycoplasma pneumoniaewere negative.Antibodies for autoimmune hepatitis and connective tissue disease (including AMA-M2,LKM-1,LC-1,SLA/LP,Ro-52,PML,sp100,gp210,M2-3E,anti-nuclear antibodies,anti-ds-DNA,Sm,SS-A,SS-B,and ENA-Jo-1) were negative.
Liver biopsy was not recommended because of the patient’s thrombocytopenia and disturbances in blood clotting functions.With written consent from her parents,genetic analysis for WD was performed.DNA was extracted from the peripheral blood samples,which were collected from the proband and her parents using the QIAamp Blood DNA Mini Kit (Qiagen,Germany).PCR was performed to amplify each exon and its neighboring introns using an ABI9700 PCR amplifier (Life Technologies,United States).Direct sequencing was performed on the amplified DNA fragments using the ABI3500 sequencer (Life Technologies,USA) and the results were subjected to sequence analysis using Sequence Scanner v1.0 (Applied Biosystems,United States).
Genetic analysis showed that the proband had a compound heterozygous mutation in theATP7Bgene;c.2333G>T (p.Arg778Leu) in exon 8 and c.3809A>G(p.Asn1270Ser) in exon 18 (reference sequence:NM_000053.3).The former was inherited from her father and the latter was inherited from her mother (Figure 1B).
The detected mutations were interpreted according to the guidelines from the American College of Medical Genetics and Genomics and patient phenotype[21].The PCR amplification and sequencing procedure were performed by Shenyang Kingmed for Clinical Laboratory (Shenyang,China),which provides third party inspection services.The hypothetical effects of the mutations on protein function were analyzed using the Polymorphism Phenotyping v2 (PolyPhen-2) prediction tool(http://genetics.bwh.harvard.edu/pph2/dbsearch.shtml),SIFT(http://sift.jcvi.org/www/SIFT_enst_submit.html),and MutationTaster(http://www.mutationtaster.org/index.html).
The c.2333G>T (p.Arg778Leu) mutation is known as a polymorphism (number rs28942074) and located in the transmembrane domain 4 (TM4).The p.Asn1270Ser mutation is located in the ATP hinge of ceruloplasmin.PolyPhen-2 and SIFT analyses suggested that the c.2333G>T mutation can negatively affect gene function,while MutationTaster suggested it is benign.PolyPhen-2,SIFT,and MutationTaster analyses suggested that the c.3809A>G mutation can be harmful (Table 1).A synonymous mutation,c.2310C>G (p.Leu770=) (rs398123136) in exon 8,was also detected in the proband and her father (data not shown).
Imaging examinations
The electroencephalogram (EEG) was normal.Brain magnetic resonance imaging scans showed a low signal intensity in T1-weighted images,and high signal intensity in T2-weighted images from the lenticular nucleus,caudate nucleus,and brainstem.Ultrasonography showed that the liver decreased in volume with an unsmooth surface and blunt edge,and the internal echogenicity was enhanced with tortuous hepatic veins.Splenomegaly was also observed.
FINAL DIAGNOSIS
WD was diagnosed.
TREATMENT
She was not treated for ITP because she had no hemorrhagic signs,but she was surveilled with the count of PLT.Liver protective therapy was begun when the patient’s liver function was found to be abnormal.After the diagnosis of WD,oral penicillamine (from 125 mg to 1000 mg per day in one week,administered in four doses finally) and zinc sulfate (300 mg per day,administered in three doses) were administered,and intramuscular injections of dimercapto propanol (100 mg per day for two weeks then weaned off over two weeks) were also given after evaluating the benefits and possible side effects.
OUTCOME AND FOLLOW-UP
The 24-h urine copper was reexamined and was 261 μg/24 h after one month.The oral therapies were continued,and dimercaptopropanol was administered for another two weeks,weaned off over two weeks,repeated over three months,and then injected once weekly.At the 6-mo follow-up,the drooling of saliva had disappeared and the tremors in the patient’s right hand and dysarthria had slightly improved,but the ultrasound results showed no marked changes.Platelet count increased and was sustained at about 60 × 109/L over the next month.
DISCUSSION
WD is an autosomal recessive disease.More than 500 mutations inATP7Bhave beenidentified in patients with WD and most patients are compound heterozygotes[6].In the proband’s family,c.2333G>T (p.Arg778Leu) and c.3809A>G (p.Asn1270Ser) were detected in the father and mother,respectively,and both mutations had been reported[13,15,22],in some cases as a compound heterozygote without thrombocytopenia[23,24].The missense heterozygous mutations in the parents were autosomal recessives,even though the mutations induced abnormal protein function according to the bioinformatic analyses.It is possible that a normal allele produces sufficient protein to transport the copper in hepatocytes.But,when the patient inherited both mutations from her parents,the deleterious effects of the compound mutation could not be counteracted by a normal allele.

Table1 Functional evaluation of the ATP7B mutations detected
WD is characterized by the toxic accumulation of copper mainly in the liver and brain,but some other systems can be involved.Hemolysis has been reported as a presenting feature in 12% of 220 WD patients[25].Thrombocytopenia with negative antiplatelet antibodies has been reported in children as a result of hypersplenism and/or a side effect of D-penicillamine therapy[26,27].It is reported that the stability of biological membranes can be disturbed by an overload of copper which was accumulated on the membranes.If the membranes of erythrocytes are affected,hemolysis can be induced.We believe that platelets are more easily destroyed and cleared in patients with WD because the membranes of platelets can also be disturbed.It has been reported that the increased depolarization of the mitochondrial membranes can enhance the apoptosis of platelets in patients with chronic ITP[28].The copper overload on membranes may also enhance apoptosis of platelets by disturbing mitochondrial membranes,but this will need to be investigated further.Serum copper is usually decreased in WD patients.Anemia and neutropenia were the most common hematologic abnormalities identified in copper deficiency patients[29,30].It was considered that hypocupremia may be a reversible cause of bone marrow dysplasia that caused cytopenia[31].Hypocupremia induced bone marrow dysplasia may be involved in thrombocytopenia.
In our case,autoimmune diseases and viral andMycoplasma pneumoniaeinfections were excluded because of the absence of autoimmune,viral,andM.pneumoniaeantibodies.Antibodies positive for GP IIb/IIIa,GP Ib/Ix,and GP Ia/type IIa and the bone marrow findings supported the diagnosis of ITP.Autoantibody-induced pathologies are quite complex[32].In our case,platelets might initially have been destroyed because of the increased copper accumulation on the membranes.The GP was then released from platelet membranes and accumulated in blood.If the autoimmune response was triggered by GP,antibodies can be generated and the platelet count can decrease further.This is a reasonable explanation for the positive GP antibodies in our case,while these antibodies were negative in the other reported cases since the cases can be at different stages of the disease.
The ultimate therapy for WD is the clearance of copper,and the prognosis is better if therapy is started as early as possible.The first-line treatment for ITP is generally using corticosteroids,or intravenous immunoglobulin in severe cases[33,34].But thrombocytopenia in WD cases has been reported to have a poor response to glucocorticoids[20,26,27].After we evaluated the possible risks to the patient,glucocorticoids were not given.The observation that the patient’s platelet counts ceased to decrease after copper clearance therapy also indicated the association of clearance of platelets with copper burden.
CONCLUSION
WD can associate with thrombocytopenia but the mechanism is still unclear.We recommend that anti-platelet autoantibodies should be tested in WD patients with thrombocytopenia in future to verify the association.
 World Journal of Clinical Cases2019年17期
World Journal of Clinical Cases2019年17期
- World Journal of Clinical Cases的其它文章
- Brachiocephalic artery stenting through the carotid artery:A case report and review of the literature
- An extremely rare pedunculated lipoma of the hypopharynx:A case report
- Calcifying fibrous tumor of the mediastinum:A case report
- Carcinoma ex pleomorphic adenoma of the trachea:A case report
- Cause of postprandial vomiting - a giant retroperitoneal ganglioneuroma enclosing large blood vessels:A case report
- Treating aplasia cutis congenita in a newborn with the combination of ionic silver dressing and moist exposed burn ointment:A case report