
Novel dual inhibitors against FP-2 and PfDHFRas potential antimalarial agents:Design,synthesis and biological evaluation
2019-02-15 02:28:38WenhuChenXueYoZhenghuiHungFeiMoLongfeiGunYunTngHulingJingJinLiJinHungLubinJingJinZhu
Chinese Chemical Letters 2019年1期
Wenhu Chen,Xue Yo,Zhenghui Hung,Fei Mo,Longfei Gun,Yun Tng,Huling Jing,Jin Li,Jin Hung,*,Lubin Jing*,Jin Zhu,*
a Shanghai Key Laboratory of New Drug Design,School of Pharmacy,East China University of Science and Technology,Shanghai 200237,China
b Key Laboratory of Molecular Virology&Immunology,Unit of Human Parasite Molecular and Cell Biology,Institute Pasteur of Shanghai,University of Chinese Academy of Science,Shanghai 200031,China
Keywords:Antimalarial drug Plasmodium falciparum FP-2 Pf DHFR Dual inhibitor
ABSTRACT Resistance to malaria parasites has quickly developed to almost all used antimalarial drugs.Cysteine protease falcipain-2(FP-2)and Plasmodium falciparum dihydrofolate reductase(PfDHFR)have crucial roles,which are absolutely necessary,in the parasite life cycle.In this study,based on the uniform pharmacophores of reported PfDHFRinhibitors and the fi rst-generation dual inhibitors against FP-2 and PfDHFR,we identi fied a novel series of dual inhibitors through fragments assembly.Lead optimization led to the identi fi cation of 14,which showed potent inhibition against FP-2 and PfDHFR enzyme(IC50=6.8?1.8 m mol/L and IC50=8.8?0.3 m mol/L)and P.falciparum 3D7 strain(IC50=2.9 m mol/L).Additionally,14 exhibited more potent inhibition to the proliferation of chloroquine-resistant P.falciparum Dd2 strain(IC50=1.1 m mol/L)than pyrimethamine(IC50>10 m mol/L),and 14 displayed micromolar inhibitory activities against two clinical isolated strains Fab9(IC50=2.6 m mol/L)and GB4(IC50=1.0 m mol/L).Collectively,these data demonstrated that 14 might be a good lead compound for the treatment of malaria.
Malaria,a mosquito-borne disease caused by infection with Plasmodium parasites,is the w orld’s most deadly parasitic infection[1,2].Almost half the w orld’s population lives in malaria endemic areas,and an estimated 1.2 billion people are at high risk of contracting the disease[3–5].According to WHO 2016,malaria caused 429,000 deaths and there were approximately 212 million clinical cases of infection globally in 2015.Since the control of malaria has been severely compromised in recent years by the w idespread resistance to nearly all frontline therapeutics which were used for both prophylaxes and treatments[3,6–8],hence,there is an urgent need for the development and discovery of new antimalarial drugs,which are structurally distinct from existing drugs and endowed with novel mechanisms of action[9].
Cysteine protease falcipain-2(FP-2)of P.falciparum is an essential hemoglobinase of erythrocytic P.falciparum trophozoites and provides the amino acids for the grow th and proliferation of Plasmodium.Many in vitro and in vivo studies have con fi rmed that inhibitors of FP-2 could block parasite hemoglobin hydrolysis,halt the development of culture parasites,and are effective against murine malaria[10,11].P.falciparum dihydrofolate reductase(PfDHFR)has received considerable attention for the prophylaxes and treatments of P.falciparum infection.Pf DHFRis one of the key enzymes in the process of DNA replication,and could catalyze 7,8-dihydrofolate to transform into tetrahydrofolate[12,13].Tetrahydrofolate serves as a necessary cofactor in the important onecarbon transfer reactions in the biosynthetic pathw ays of pyrimidines,purines,and amino acids[14].Thus,a more powerful pesticidal effect could be achieved by inhibiting FP-2 and PfDHFR simultaneously.Such dual inhibitorsmight show a good synergetic effect,and overcome the drug-resistance and be capable of providing “a combination therapy”in a single agent[15].
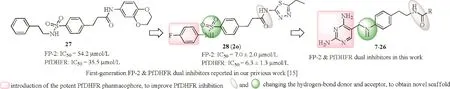
Fig.1.Chemical modification strategies for the second-generation dual inhibitors.(For interpretation of the references to color in the text,the reader is referred to the web version of this article.)
Previously,we reported the fi rst-generation dual inhibitors against FP-2 and Pf DHFR based on the compound 27,which was randomly identi fied by screening FP-2 inhibitors in our laboratory,and gained compound 2o(28)which exhibited a high enzymatic inhibition and a moderate in vivo antimalarial ef fi cacy[15](Fig.1).Based on the SARof the fi rst-generation dual inhibitors[15],to gain new scaffold dual inhibitors with more potencies,a novel series of dual inhibitors were designed,synthesized by displacing 4-fl uorophenyl with the uniform pharmacophores of reported Pf DHFR inhibitors,2,4-diamino heterocyclic fragments(Table S1 in Supporting information).Considering the feasibility of the synthesis and novelty of scaffold,the sulfonamide was displaced with the secondary amine(green,Fig.1),and the amide position was interchanged(gray,Fig.1).Therefore,2,4-diaminopyrimidine analogues with substituent(R,Fig.1)at the terminal amide(7–26)were synthesized to obtain better dual target inhibitors and explore the SAR.
First of all,we synthesized all the 2,4-diaminopyrimidine analogues(7–26)as described in Scheme 1.2,4-Diaminopyrimidine-5-carbonitrile(2)was prepared by reaction of guanidine carbonate(1)with ethoxymethylenem alononitrile in EtONa and EtOH in a 71%yield.Further treatment of 2 with Nickel in MeOH under H2for 24 h gave rise to 2,4-diaminopyrimidine-5-carbaldehyde(3)in an 80%yield.Subsequent condensation of commercially available 4-nitrophenethylamine hydrochloride(4)with appropriate acids RCOOH in the presence of HOBt,EDCIand DIPEA in N,N-dimethylacetamide afforded N-(4-nitrophenethyl)amide 5 in good yields.Reduction of 5 in the presence of 10%Pd/Cand H2in MeOH overnight in good yields provided N-(4-aminophenethyl)amide(6).Analogues 7–26 were performed by the reaction of 3 and 6,via the condition using NaCNBH3in MeOH under re fl ux overnight in good yields(30%–70%)and were characterized by1H NMR and HRMS.The purity was over 95%,as determined by HPLCanalysis(Table S2 in Supporting information).Supplementary data associated with experiment section,reported Pf DHFR inhibitors and HPLC analysis data of compounds 7–26 can be found in Supporting information.
Then the twenty analogues were evaluated for FP-2 enzymatic inhibitory activity and for PfDHFR enzymatic inhibitory activity using the cysteine protease inhibitor(E-64)and pyrimethamine as the reference standard in the assay,respectively.And we assessed the inhibition rate(IR)of all the analogues against FP-2 and Pf DHFR at 10 m mol/L fi rstly.A set of analogues with various substituents(R),including electron-donating substituted phenyl ring(9–13),electron-withdraw ing substituted phenyl ring(14–17),hetero-aryl rings(18–22)and alkyl groups(23–26),were evaluated for IR against FP-2 and Pf DHFR,respectively.From the resultsin Table 1,3 analogues,i.e.,11,14 and 15,were identi fied as inhibitors against FP-2 with IR>40%at 10 m mol/L and 19 analogues,i.e.,7–24,26,were identi fied as potent inhibitors against Pf DHFRwith IR>70%at 10 m mol/L.Therefore,the IC50value of them for the enzymatic inhibitory activity was further evaluated.
Analysisof the data show n in Table 1 revealed some notew orthy observations from the SAR study of analogues 7–26:(1)The inhibitory activities against FP-2 showed the Rsubstituents which were phenyl rings with electron-donating groups(EDGs)preferred 3-position(9 vs.10 and 11 vs.12)and the Rsubstituents which were phenyl rings with electron-withdraw ing groups(EWGs)favored 4-position(14 vs.15 vs.16);(2)In the studied sets of the R substituents,the potency against FP-2 substantially increased in the order of EWG-aryl>EDG-aryl>(aryl)alkyl;(3)In the test of inhibitory activities against PfDHFR,the R substituents could be well tolerated and 19 analogues displayed high potencies(IR at 10 m mol/L>70%);the potency against Pf DHFR substantially increased in the order of EDG-aryl>(aryl)alkyl>EWG-aryl.
From the results described above,analogue 14 was identi fied as a potent inhibitor against both FP-2 and Pf DHFR(IC50=6.8 m mol/L against FP-2,IC50=8.8 m mol/L against PfDHFR).Therefore,analogue 14 and the lead compound 28 were next evaluated in the inhibitory activity against the blood stage of the multi-drugsensitive P.falciparum 3D7 strain and P.falciparum Dd2 strain,which carry a phenotype of resistance to chloroquine[16,17].Compound 28 displayed poor potencies against 3D7 strain(IR=7.9%@20 m mol/L)and Dd2 strain(IR=6.2%@20 m mol/L).Asshow n in Fig.S1(Supporting information),analogue 14 together with artemisinin(Art)were tested in 3D7 cells.Analogue 14 performed micromolar potencies against 3D7 parasites(IC50=2.9 m mol/L).Moreover,as show n in Fig.S2(Supporting information),14 displayed micromolar potency against Dd2(IC50=1.1 m mol/L)while pyrimethamine presented less effective inhibition against Dd2(IC50>10 m mol/L),which indicated analogue 14 could also inhibit the grow th of resistant P.falciparum.Furthermore,we evaluated the inhibitory activities of compound 14 against tw o clinical isolated strains,Fab9 and GB4.To our delight,as show n in Fig.S3(Supporting information),14 also displayed micromolar potencies against both strains(IC50=2.6 m mol/L against Fab9 and IC50=1.0 m mol/L against GB4).
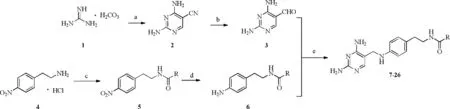
Schem e 1.Synthesis of the 2,4-diaminopyrimidine analogues(7-26).Reagents and conditions:(a)Ethoxymethylenemalononitrile,Et ONa/EtOH,5?C,8 h;(b)Ni/H2,MeOH,25?C,24 h;(c)HOBt,EDCI,DIPEA,RCOOH,DMA,25?C,overnight;(d)10%Pd/C,H2,MeOH,re fl ux;(e)NaCNBH3,MeOH,re fl ux,overnight.

Table 1(Continued)
Moreover,to understand the structural basis for the inhibitory activities of the inhibitors against FP-2 and PfDHFR,the 3Dbinding models of analogue 14 with FP-2 and PfDHFR were studied by molecular docking(Fig.2).Fig.2A show s the predicted binding poses of 14 in the catalytic site of FP-2.The amide moiety of 14 directly interacted with Asn156 via H-bonds,Meanwhile,the terminal 2,4-diaminopyrimidine group and secondary amine interacted with Asp18 and Lys20 via H-bonds.For PfDHFR(Fig.2B),the catalytic subpocket formed around Asp54 were occupied by the 2,4-diaminopyrimidine group in 14.The 2,4-diaminopyrimidine ring formed a complicated hydrogen-bond network with Asp54,Ile14,and Ile164,respectively,which contributed significantly to the higher af fi nity of 14.Moreover,the amide group formed an H-bond with the key residue Arg122.
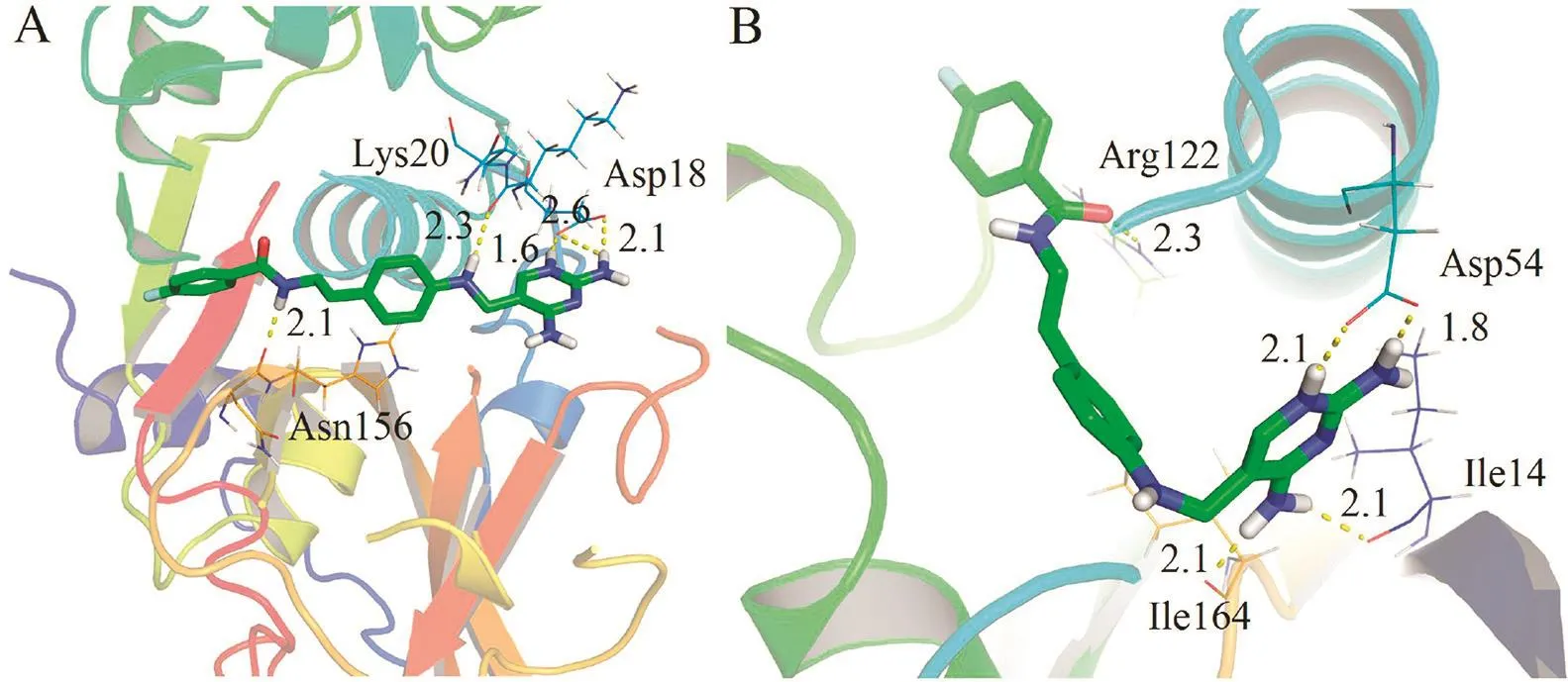
Fig.2.Molecular docking studies on 14.Docked poses of 14(green sticks)in the active sites of FP-2(A)and PfDHFR(B),respectively.Key residues of the binding pockets are show n as lines.Hydrogen bonds are show n with yellow dash lines.
In summary,we have identi fied a novel series of 2,4-diaminopyrimidine analogues(7–26)derived from 28 as FP-2 and PfDHFRdual inhibitors.On the basisof the structure of the lead compound 28,20 novel 2,4-diaminopyrimidine analogues have been synthesized and tested in FP-2 and Pf DHFR enzymatic inhibitory activity assays.Molecular docking studies showed that the amide and secondary amine groups of new analogues could form hydrogen bonds with the surrounding amino acid residues,which were the same as the fi rst generation dual inhibitors[15],combining with the preliminary SARs,we speculated the amide and secondary amine groups inherited FP-2 inhibitory activity from 28 and 2,4-diaminopyrimidine groups contributed to the inhibitory activity against PfDHFR.The in vitro inhibitory activity against P.falciparum assays with multi-drug-sensitive strain 3D7 further con fi rmed that analogue 14 was a good parasites inhibitor(IC50=2.9 m mol/L).To our delight,the inhibition of 14 against chloroquine-resistant Plasmodium falciparum strain Dd2(IC50=1.1 m mol/L)was more superior than pyrimethamine(IC50>10 m mol/L)and analogue 14 displayed micromolar inhibitory activities against tw o clinical isolated strains Fab9(IC50=2.6 m mol/L)and GB4(IC50=1.0 m mol/L).Overall,14 has the potential to be developed as a lead compound of antimalarial drugs.
Acknow ledgm ents
Financial support for this research provided by the National Natural Science Foundation of China(Nos.21372001 and 21672064),the “Shu Guang”Project supported by the Shanghai Municipal Education Commission and Shanghai Education Development Foundation(No.14SG28),and the Fundamental Research Funds for the Central Universities are gratefully acknow ledged.
Appendix A.Supplem entary data
Supplementary data associated with thisarticle can be found,in the online version,at https://doi.org/10.1016/j.cclet.2017.11.041.
 Chinese Chemical Letters2019年1期
Chinese Chemical Letters2019年1期
- Chinese Chemical Letters的其它文章
- Information for authors
- A one-pot protocol for copper-mediated azide–alkyne cycloaddition using alkenyl tri fl ate precursors
- One-pot synthesis of tetrahydroindoles via a copper catalyzed N-alkynation/[4+2]cycloaddition cascade
- Gram-scale preparation of dialkylideneacetones through Ca(OH)2-catalyzed Claisen-Schmidt condensation in dilute aqueous EtOH
- Tetra-phthalimide end-fused bi fl uorenylidene:Synthesis and characterization
- Water bridges are essential to neonicotinoids:Insights from synthesis,bioassay and molecular modelling studies