
鎳催化芳烴鹵化物還原性交叉偶聯(lián)的反應(yīng)機(jī)理
2014-06-23 06:51:48任清華
物理化學(xué)學(xué)報(bào) 2014年5期
蔣 峰 任清華
(上海大學(xué)理學(xué)院化學(xué)系,上海200444)
1 Introduction
Unsymmetrical biaryl compounds are very important intermediates in modern organic synthesis and exist extensively in biologically active molecules.1The unsymmetrical biaryl units play a major role in natural product synthesis,2,3organic semiconductor,4material science,5,6and drug design.7In the last several decades,a lot of methods based on microwave,8,9photochemistry,10or other approaches11,12have been developed to form biaryl compounds.Among these methods,transition metal catalyzed cross-coupling reactions are important strategies,13-15especially in the formation of unsymmetrical biaryls.16-19
Unfortunately,most of these reactions involve air-and moisture-sensitive organometallic reagents,which often cause some troublesome problems such asβ-H elimination and slow reductive elimination.20,21Therefore much attention has been paid to the reductive cross-coupling of aryl halides to form the biaryls.For example,Amatore and Gosmini22have studied Co-catalyzed formation of unsymmetrical biaryls and Gonget al.23have reported Ni-catalyzed cross-coupling of aryl halides to form unsymmetrical biaryls.
However,although transition metal catalyzed reductive crosscoupling reactions have been widely studied experimentally,no theoretical investigation of the mechanisms of Ni-catalyzed reductive cross-coupling of aryl halides to form unsymmetrical biaryl using density functional theory(DFT)method has yet been reported.
In this paper,we explore the Ni-catalyzed formation of unsymmetrical biaryls from bromobenzene(R1)and methyl 4-bromobenzoate(R2)using the DFT method.We mainly try to investigate the following questions.Firstly,how does the overall catalytic cycle take place?Secondly,are organometallic reagents formed in the reaction process?And finally,which reactant is favored to combine with the active Ni catalyst during the formation of the unsymmetrical biaryl?
Our model calculations are based on the experimental work ofGong′sgroup.23The scheme of the reaction is shown in Fig.1.In the experimental study,R1and R2were the reactants and 4,4'-di-methyl-2,2'-bipyridine was the ligand.NiBr2was used as the catalyst precursor and CH3CN as the solvent.
2 Computational details
All calculations were implemented in the Gaussian 03 program24using density functional theory method with the B3LYP hybrid functional.25-28The 6-31G*basis set was used for C,H,O,and N,and the 6-311G*basis set was used for the Br atom.29,30The SDD quasi-relativistic pseudopotential and associated basis set was employed for Ni.31All gas phase geometries were fully optimized without any symmetry restriction,following the vibrational frequencies analysis to ensure that the local minima had zero imaginary frequencies and the transition state had exactly one.The C-PCM(conductor-like PCM)32single point calculation with the UFF radii based on the gas-phase optimized structure was carried out to evaluate the solvation effect in CH3CN solvent.Both the electronic and nonelectronic free energies in solution were added to the gas-phase Gibbs free energies to obtain the solution Gibbs free energies in CH3CN(ΔGsol,298 K).
3 Results and discussion
The investigations of the mechanisms for both Ni0-catalyzed33,34and NiI-catalyzed35-37processes have attracted considerable attention in recent years.Based on these previous works,we propose that the mechanisms of the reductive crosscoupling reaction shown in Fig.1 include the following basic steps:(1)first oxidative addition,(2)reduction,(3)second oxidative addition,(4)reductive elimination and catalyst regeneration.The detailed catalytic cycles are outlined in Fig.2.
3.1 First oxidative addition
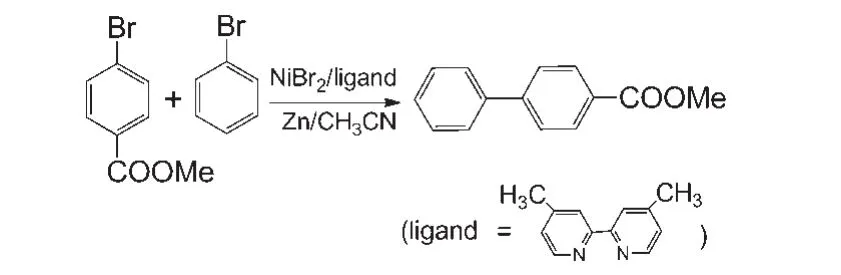
Fig.1 Ni-catalyzed reductive cross-coupling reaction of bromobenzene and methyl 4-bromobenzoate
It can be seen from Fig.2 that the starting species NiBr2is reduced by Zn while connecting with the ligand to form the starting catalyst CA1,NiI-L-Br.The reaction goes through Path II if the reactant R1firstly attacks CA1,while Path IV labels the mechanism in which the reactant R2first attacks CA1.However,it is also possible that the catalyst CA1is reduced by Zn again to form the active Ni0catalyst CA0.Then the reactant R1attacks CA0to go through Path I or the reactant R2attacks CA0to go through Path III.We explore all possible paths below.
3.1.1 First oxidative addition catalyzed by Ni0(Path I and Path III)
As far as what we know,Ni0and NiIIcompounds have two multiplicities,namely,in the singlet or triplet states.Both of them will be discussed in the following studies.
3.1.1.1 First oxidative addition of triplet Ni0with the reactants(R1in Path I and R2in Path III)
For the studied system shown in Fig.1,the single electron transfer mechanism could not be found although it has been reported in the process of oxidative addition of the triplet Ni complexes with organohalogen compounds.38,39In the triplet Ni0mechanism of Path I or Path III,aπcomplex is formed before the first oxidative addition aiming to minimize the system energy,which is shown in Fig.3.The approach of R1or R2towards the active catalyst triplet Ni0(L),CA0t,leads to the formation ofπcomplexes(CP1tor CP2t),which decreases the electronic energy by 65.52 or 72.05 kJ·mol-1,respectively.
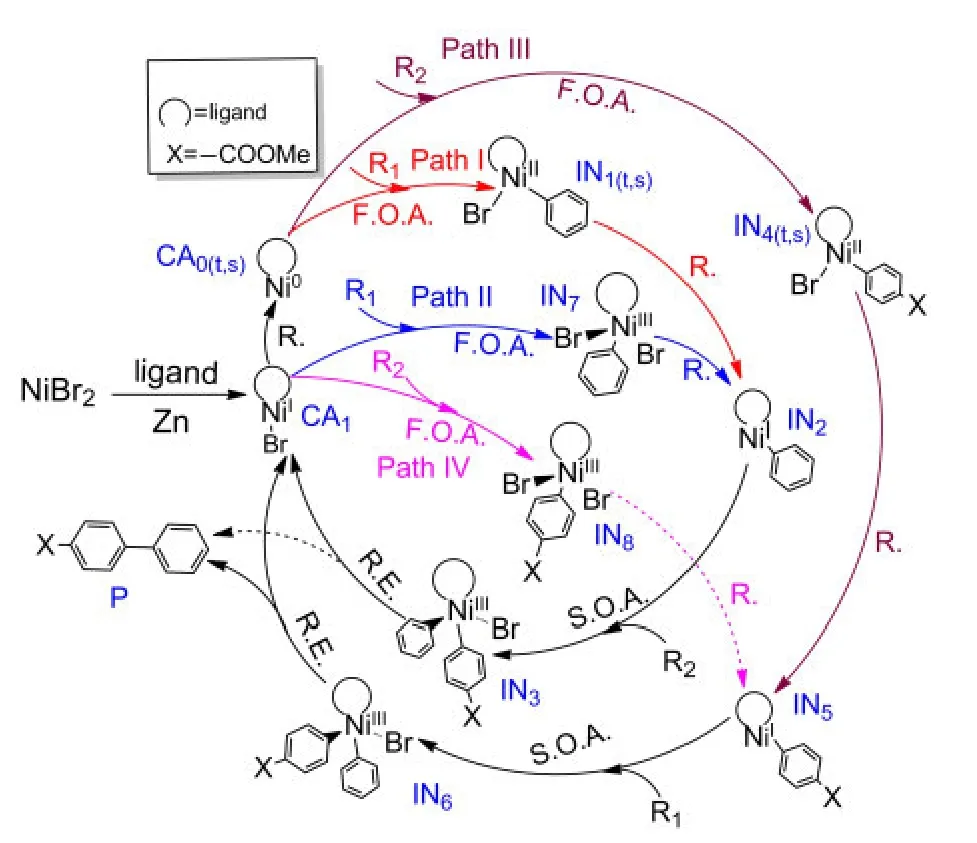
Fig.2 Outline of the mechanisms for Ni-catalyzed formation of unsymmetrical biaryl Pfrom R1and R2
From CP1t(triplet Ni0)or CP2t(triplet Ni0),after passing through the transition state TS1t(triplet Ni0)or TS4t(triplet Ni0),the intermediate IN1s(singlet NiII)or IN4s(singlet NiII)is formed.It is probably that the TS1tor TS4ttransforms adiabatically into IN1sor IN4sfrom CP1tor CP2tby a spin intercrossing process with a singlet potential energy surface.40The energy of IN1s(singlet NiII)or IN4s(singlet NiII)is 18.68 or 20.22 kJ·mol-1smaller than IN1t(triplet NiII)or IN4t(triplet NiII),which means that the singlet intermediate IN1sor IN4sis more stable.From the optimized structures shown in Fig.3,it can be seen that the Br,C,and two N atoms around the Ni center in TS1tand IN1sare nearly in one plane,but the Br―Ni bond in IN1tis nearly perpendicular to the plane of N―Ni―N.The optimized structure of IN1tor IN4t(shown in the Supporting Information)is very different from that of TS1tor TS4t.Nevertheless,it is also possible that the system proceeds from CP1tor CP2tto IN1tor IN4trather than to IN1sor IN4sconsidering the fact that the energy differences between the singlet and the triplet intermediates are not too much.
From the optimized structures shown in Fig.3,it can be seen that the distance of Br-Ni in CP1tis 0.315 nm.It becomes shorten to 0.292 nm in TS1tand to 0.233 nm in IN1s.At the same time the Br―C bond is broken in IN1s(0.297 nm).From Fig.3,we can also see that the energy barrier for the first oxidative addition of the triplet Ni0-catalyzed step in Path I or in Path III is very small(9.42 kJ·mol-1for TS1t,5.35 kJ·mol-1for TS4t).
3.1.1.2 First oxidative addition of singlet Ni0with the reactants(R1in Path I and R2in Path III)
The energy of the singlet Ni0catalyst CA0s(shown in Fig.4)is 82.35 kJ·mol-1higher than the triplet Ni0catalyst CA0t.The detailed data of them are listed in the Supporting Information.The possible reason is that CA0spresents a distorted structure(the dihedral angle N―C―C―N is 25.5°),while CA0thas an N―C―C―N planar structure.The transition state from CP1sor CP2sto attain the intermediate IN1sor IN4sis TS1sor TS4s,respectively,where the corresponding electronic energy barrier is 39.77 or 32.62 kJ·mol-1.From the optimized structures shown in Fig.4,it can be seen that the Br―C bond is broken in IN1s(0.297 nm,compared to 0.203 nm in CP1sand 0.195 nm in TS1s),while the Ni―C bond is formed in IN1s(0.189 nm,compared to 0.282 nm in TS1sand 0.357 nm in CP1s).The process in going from CP2sto obtain IN4sis similar.From Fig.3 and Fig.4,we can see that the energy barrier of the singlet Ni0-catalyzed step(39.77 kJ·mol-1for TS1sin Path I or 32.62 kJ·mol-1for TS4sin Path III)is higher than that of the triplet Ni0-catalyzed step(9.42 kJ·mol-1for TS1tin Path I or 5.35 kJ·mol-1for TS4tin Path III)in the first oxidative addition,which means that the triplet Ni0mechanism is favored over the singlet Ni0mechanism.
3.1.2 First oxidative addition catalyzed by NiI(Path II and Path IV)
The NiI-catalyzed process starts from the active catalyst[NiI(L)(Br)],CA1.No complex could be found when the catalyst CA1attacks the reactant R1or R2.For a similar situation,Cárdenaset al.41also could not find a complex structure when they studied the mechanism of Ni-catalyzed cross-coupling of alkyl zinc halides for the formation of C(sp2)―C(sp3)bonds.The energy profiles and the optimized geometries of the NiI-catalyzed processes are shown in Fig.5.The intermediate IN7(for Path II)or IN8(for Path IV)is obtained from the oxidative addition of CA1with R1or R2after passing through the transition state TS7or TS8,respectively.
In the first oxidative addition step of NiI-catalyzed process,it can be seen that the energy barrier of TS7is 53.34 kJ·mol-1for Path II,while the energy barrier of TS8is 45.72 kJ·mol-1for Path IV.Obviously,they are much higher than the energy barrier of TS1t(9.42 kJ·mol-1)or TS4t(5.35 kJ·mol-1)for the triplet Ni0-catalyzed processes.Overall,the triplet Ni0-catalyzed mechanism is favored for the first oxidative addition.
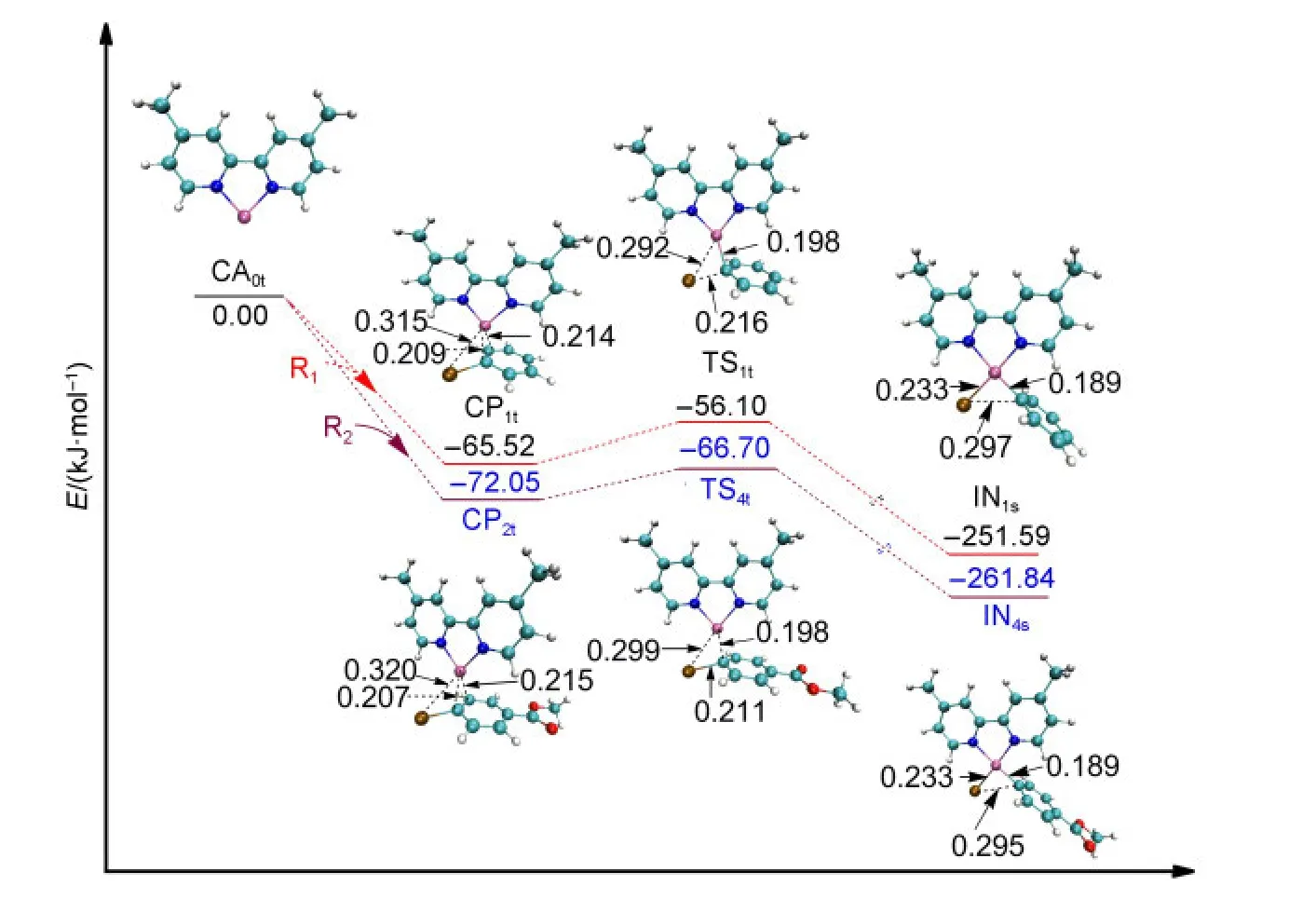
Fig.3 Optimized structures and electronic energies of the first oxidative addition of triplet Ni0catalyst(CA0t)with the reactant R1or R2

Fig.4 Optimized structures and electronic energies of the first oxidative addition of singlet Ni0catalyst(CA0s)with the reactant R1or R2
3.2 Reduction
The reduction processes by Zn powder in this article were not computed based on the following reasons:currently,no suitable model can be used to describe Zn aggregates;furthermore,the barrier for the reduction step is negligible compared to the energy of the rate-determining step.42The reduction step of a catalyst precursor to produce the catalyst is the first step in many traditional cross-coupling reactions,which is generally ignored in studies of the mechanism of transition metal catalyzed cross-coupling reactions.35,41,43Hence,it is safe to ignore these processes for our system.The middle product of reduction from IN1(t,s)for Path I or from IN7for Path II is IN2and the middle product of reduction from IN4(t,s)for Path III or IN8for Path IV is IN5.
3.3 Second oxidative addition
The second oxidative addition is initiated from the middle product of reduction IN2or IN5.The approach of R2towards IN2leads to the formation of the intermediate IN3after passing through the transition state TS2.The energy barrier of TS2is 70.50 kJ·mol-1.Similarly,the approach of R1towards IN5results in the formation of the intermediate IN6after passing through the transition state TS5with the energy barrier of 49.66 kJ·mol-1.All of the optimized geometries and the electronic en-ergies are shown in Fig.6.It can be seen that the bond of Ni atom with the2C atom which came from R2is formed in IN3(0.194 nm,compared to 0.208 nm in TS2).Similarly,the bond of Ni atom with the1C atom coming from R1is formed in IN6(0.195 nm,compared to 0.212 nm in TS5).We can also see that the energy values of the transition states TS2and TS5are close and their optimized structures are similar.From the orbital analyses of TS2and TS5,their HOMO orbitals are also very similar(shown in the Supporting Information).These results show that it makes no significant difference to the second oxidative addition step whether R1or R2is the first to attack the catalyst.
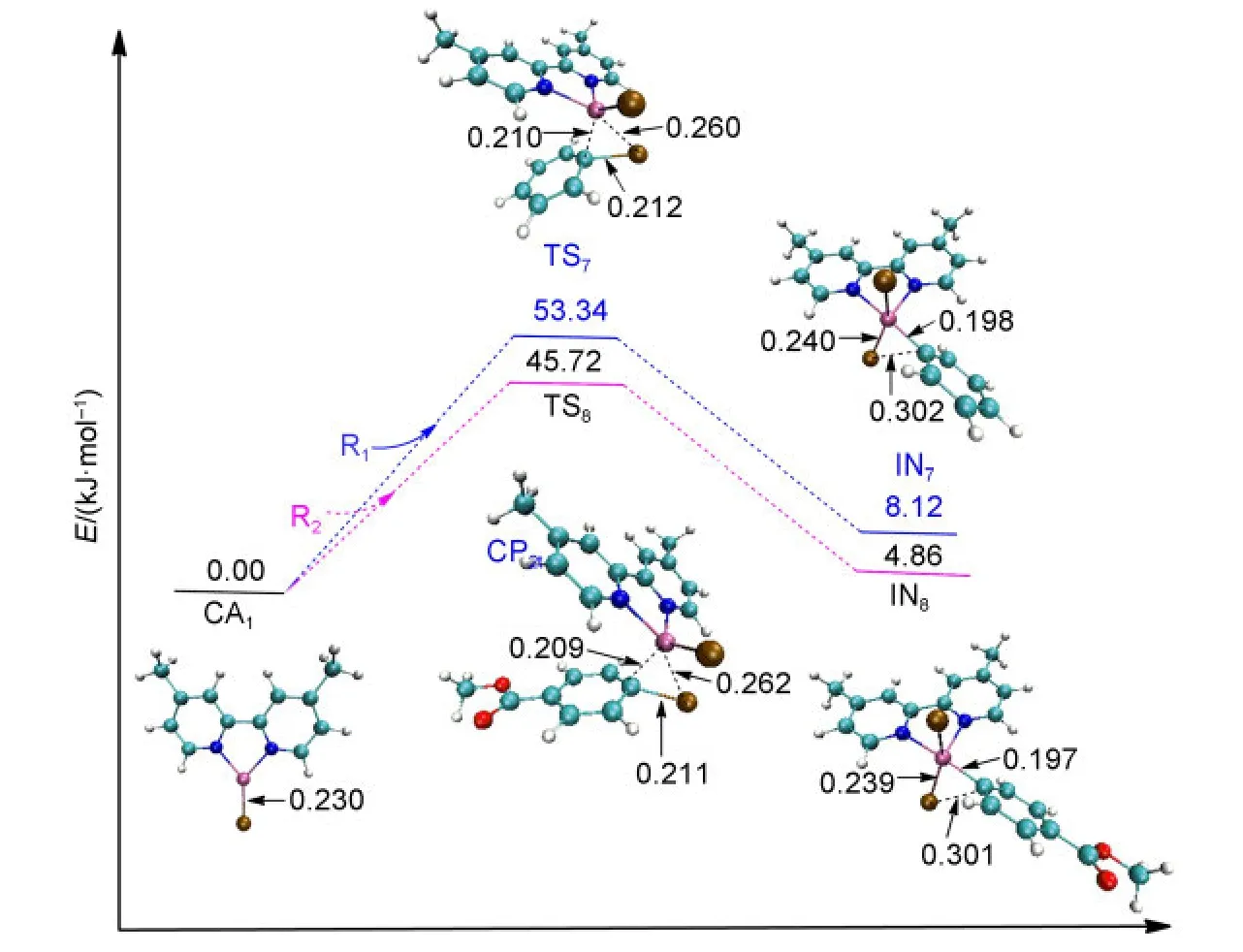
Fig.5 Optimized structures and electronic energies of the first oxidative addition of NiIcatalyst(CA1)with the reactant R1or R2
3.4 Reductive elimination
Once the intermediate of the second oxidative addition IN3or IN6has been produced,the reductive elimination step is an easy process without any large energy barriers.The barrier for passing through the transition state TS3from IN3to obtain the final product P while regenerating the catalyst CA1is 11.09 kJ·mol-1.Similarly,the energy barrier for passing through the transition state TS6from IN6to regenerate the catalyst CA1is 9.01 kJ·mol-1(see Fig.7).The values of the energy barriers for the reductive elimination steps are very small and our calculated results are in agreement with the work of Liuet al.43From the optimized structures shown in Fig.7,the C―C bond is formed in the final product P(0.148 nm,compared that the distance of C―C is 0.213 nm in TS3and 0.211 nm in TS6).
3.5 Comparison of the catalytic cycles
Based on the above results,we can see that the triplet Ni0-catalyzed mechanism is favored over both the singlet Ni0-catalyzed mechanism and the NiI-catalyzed mechanism for the reaction system shown in Fig.1.The favored triplet Ni0-catalyzed mechanism involves Path I where the Ni0catalyst first combines with R1or Path III where the Ni0catalyst first attacks R2.The energy profiles for the whole catalytic cycles of Path I and Path III are listed in Fig.8.It can be seen that the rate-determining step for Path I or Path III is the second oxidative addition step,where the energy barrier is 70.50 kJ·mol-1for Path I and 49.66 kJ·mol-1for Path III,respectively.The difference is small,which means that the preferential combinations of the active catalyst with the substrate R1or R2are both possible in this reaction system.Also,the reaction is produced at room temperature,23so the values of the energy barriers for the triplet Ni0mechanism are reasonable.
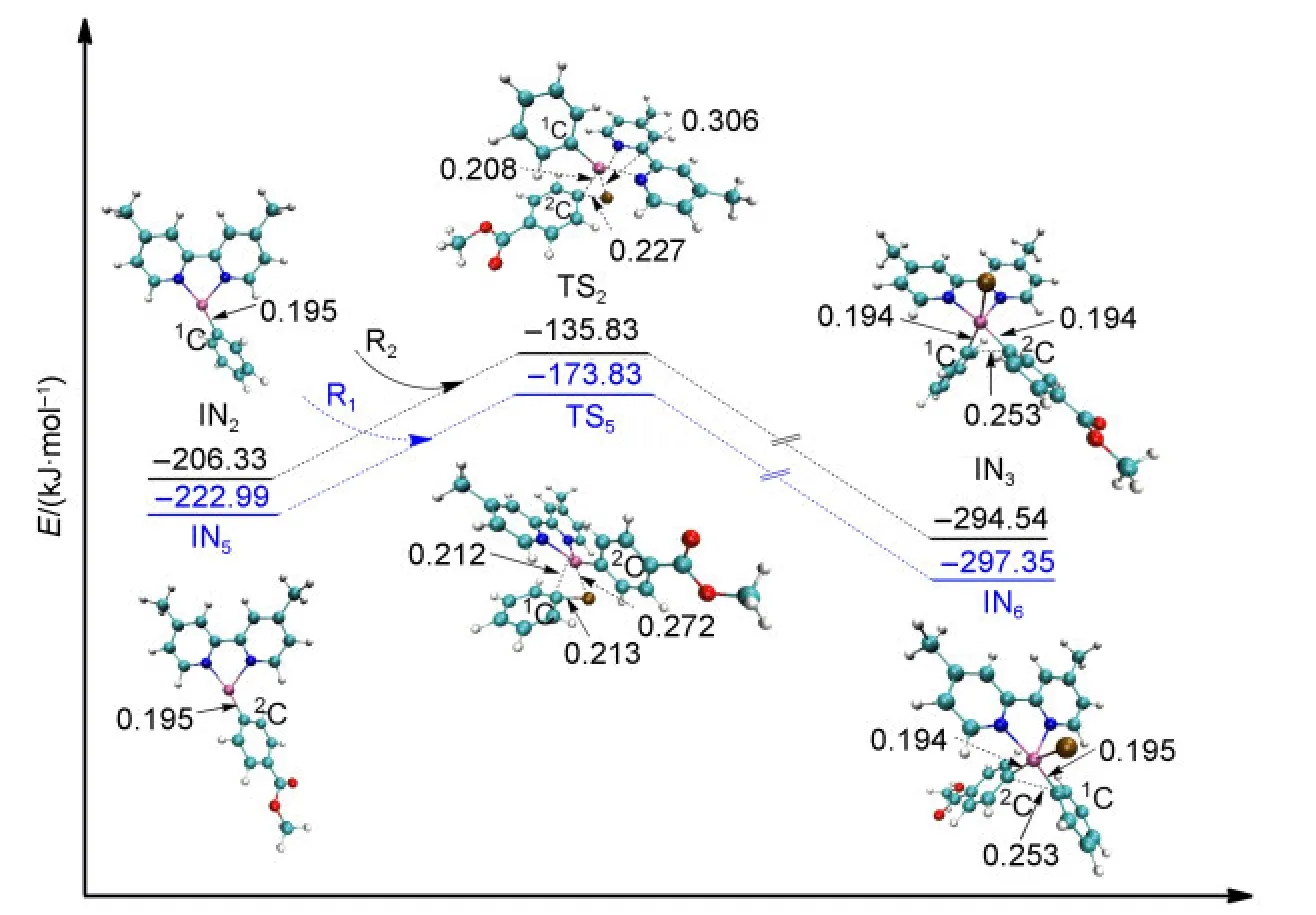
Fig.6 Optimized structures and electronic energies of the second oxidative addition of IN2and IN5with the reactant R1or R2
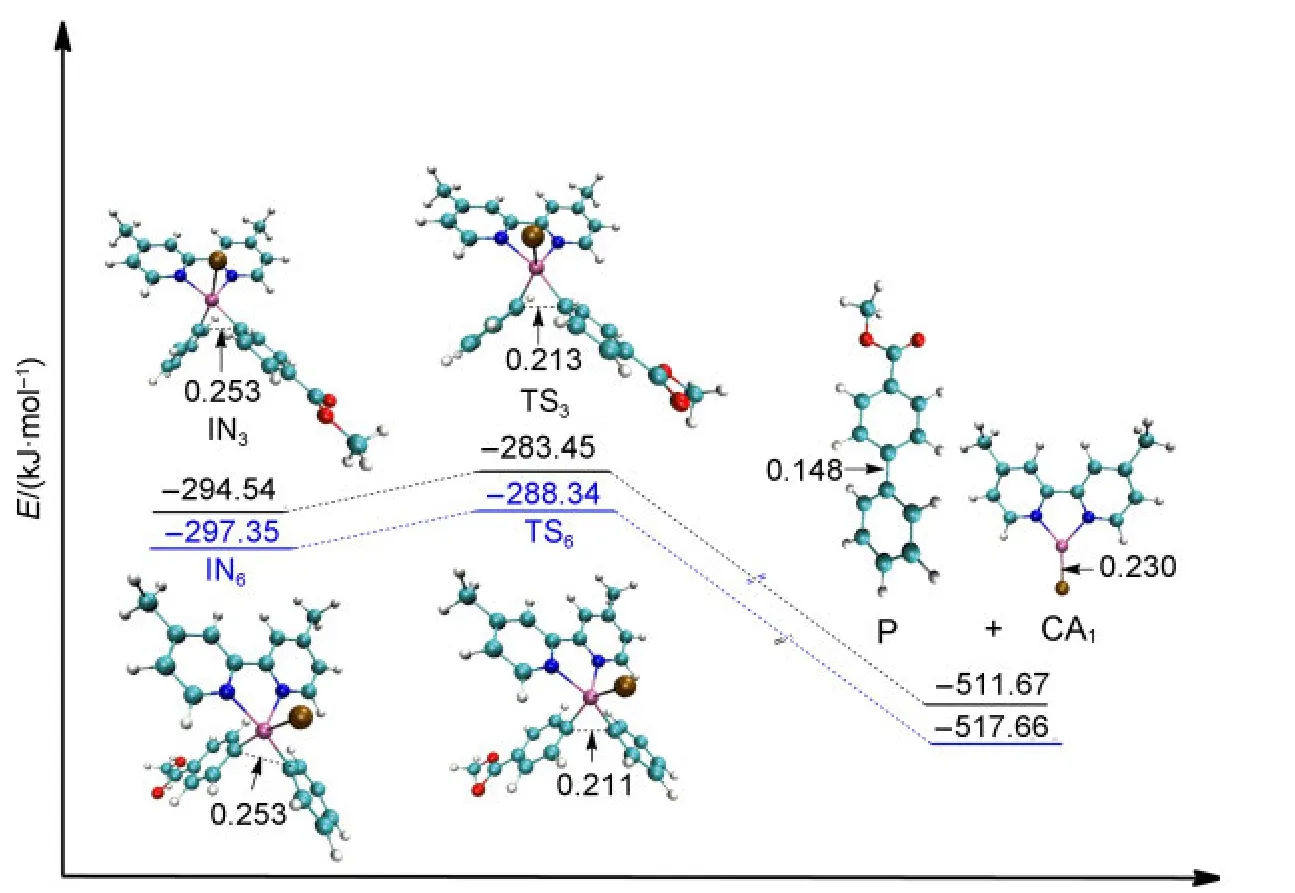
Fig.7 Optimized structures and electronic energies of reductive elimination
3.6 Solvent effect
All of the above results are calculated in gas phase.In order to investigate whether solvent effects can play a significant role in the process,we discuss here the calculated results for the Gibbs free energies in CH3CN solution for Path I.The calculated results for Path III are listed in the Supporting Information.
The Gibbs free energies in gas phase(ΔG)and the Gibbs free energies in CH3CN solvent(ΔGsol,298 K)for the overall catalytic cycles of Path I are shown in Fig.9.The former is depicted as a solid line and the latter is drawn as a dashed line.It can be seen that the solvent effect is small in the first oxidative addition step.However,the solvent effect is obvious for the second oxidative addition step,where the ΔGsolis 86.04 kJ·mol-1,compared to the ΔG,123.70 kJ·mol-1.The solvent effect leads to a lowering of the energy barrier for the rate-limiting step in CH3CN solution.For the next reductive elimination step,the solvent effect is not significant.The ΔGsolis 11.64 kJ·mol-1,compared to the ΔG,7.87 kJ·mol-1.
Overall the rate-determining step in solution is still the second oxidative addition and the solvent effect does not change the mechanism of the reaction.The results obtained from the gas-phase calculations can effectively be used for the reaction.
3.7 Organometallic reagents
In the last several years,transition metal catalyzedin situKumada and Negishi cross-coupling reactions have been ex-tensively studied.For instance,Czaplik et al.44investigated Fe-catalyzed Kumada cross-coupling reactions which need not prepare organomagnesium species;Co-catalyzed Negishi crosscoupling reactions of arylzinc species with benzyl chlorides were explored by Amatore and Gosmini;45Lipshutzet al.46investigated the Pd-catalyzed formation of unsymmetrical diarylmethanes without preparation of organozinc reagents.All of these outstanding works lead us to consider whether organometallic reagents are or are not produced in the reductive crosscoupling reaction of aryl halides.In order to answer this question,we simulate the process of the formation of arylzinc species in our reaction system.As shown in Fig.10(a),the energy barrier for passing through the transition state TS9to produce the organozinc reagent IN9in the oxidative addition step of Zn with reactant R1is 177.44 kJ·mol-1.This means that the organozinc reagent IN9is unlikely to be obtained in the mild experimental reaction condition.23Similarly,the energy barrier of TS10is 176.60 kJ·mol-1in Fig.10(b).This also suggests that the organozinc reagent IN10cannot be formed in our reaction condition.Our simulated results are in keeping line with the experimental data of Ni-catalyzed cross-coupling of aryl halides and alky halides inWeix′sgroup.47
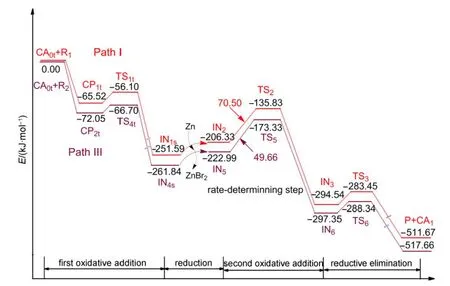
Fig.8 Energy profiles for the overall catalytic cycles of Path I and Path III
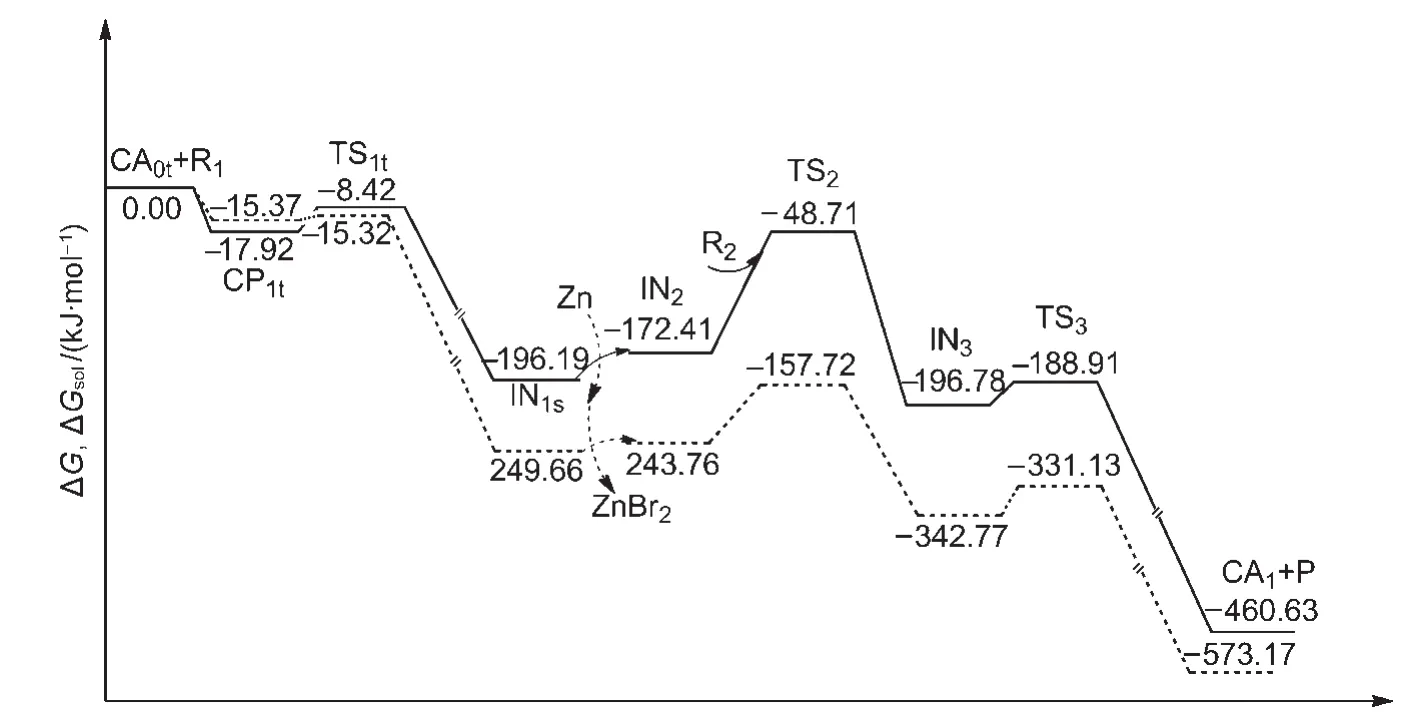
Fig.9 Gibbs free energies in gas phase(ΔG,solid line)and Gibbs free energies in solvent CH3CN(ΔGsol,298 K,dashed line)for the overall catalytic cycles of Path I
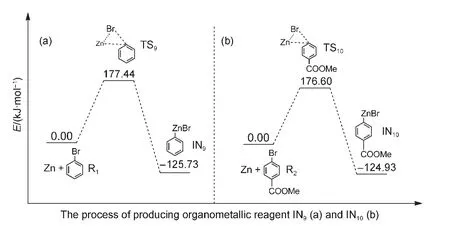
Fig.10 Energy profiles for the processes of producing the possible organometallic reagents
4 Conclusions
In this paper,DFT calculations using the B3LYP method have been carried out to thoroughly explore the mechanisms of the Ni-catalyzed reductive cross-coupling reaction of bromobenzene(R1)and methyl 4-bromobenzoate(R2)to form an unsymmetrical biaryl.
Our calculated results showed that the favored mechanism is the triplet Ni0acting as the active catalyst,and includes four basic steps:(1)first oxidative addition;(2)reduction;(3)second oxidative addition;(4)reductive elimination and regeneration of the catalyst.Whether the triplet Ni0catalyst combines initially with the reactant R1or R2has a small effect on the overall catalytic cycle.When the active Ni0catalyst firstly combines with R1in Path I,the energy barrier for the rate-limiting step is 70.50 kJ·mol-1,which is a little higher than that for the Ni0catalyst combining first with R2in Path III(49.66 kJ·mol-1).Additionally,the energy barriers of producing organozinc reagents IN9and IN10are 177.44 and 176.60 kJ·mol-1,respectively,which means that organozinc reagents are extremely difficult to produce under the mild experimental reaction conditions which have actually been used.
Homocoupling acting as a side reaction is always present in the transition metal catalyzed cross-coupling reactions.As far as we know,the experimental yield of the cross-coupling reaction of R1and R2is 52%.23Hence,the homocoupling byproducts is also expected to exist in this kind of reaction.Our calculated results show that the energy barriers of generating Ph―Ph or MeCOO―Ph―Ph―COOMe are 49.83 and 40.45 kJ·mol-1,respectively(the detailed data are listed in the Supporting Information).This proves that the homocoupling reactions are possible.Our recently paper48has already explored the mechanisms of homocoupling to form biphenyl in a similar system.So we have not discussed it here.
Supporting Information:Detailed molecule coordinates of all optimized structures,the calculated energy values,the optimized structures of IN1tand IN4t,the HOMO orbitals of TS2and TS5,Gibbs free energies ΔGand ΔGsolof Path III have been included.This information is available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
(1) Torssell,K.B.Natural Product Chemistry:a Mechanistic and Biosynthetic Approach to Secondary Metabolism;John Wiley&Sons:New Jersey,1983;pp 401-404.
(2) Bonesi,S.M.;Fagnoni,M.;Albini,A.Angew.Chem.Int.Edit.2008,47,10022.doi:10.1002/anie.v47:52
(3) Corbet,J.P.;Mignani,G.Chem.Rev.2006,106,2651.doi:10.1021/cr0505268
(4) Roncali,J.Chem.Rev.1992,92,711.doi:10.1021/cr00012a009
(5)Yang,W.Y.;Ahn,J.H.;Yoo,Y.S.;Oh,N.K.;Lee,M.Nat.Mater.2005,4,399.doi:10.1038/nmat1373
(6)Huang,Z.;Lee,H.;Lee,E.;Kang,S.K.;Nam,J.M.;Lee,M.Nat.Commun.2011,2,459.doi:10.1038/ncomms1465
(7) Hajduk,P.J.;Bures,M.;Praestgaard,J.;Fesik,S.W.J.Med.Chem.2000,43,3443.doi:10.1021/jm000164q
(8) Larhed,M.;Hallberg,A.J.Org.Chem.1996,61,9582.doi:10.1021/jo9612990
(9) Blettner,C.G.;Ko¨nig,W.A.;Stenzel,W.;Schotten,T.J.Org.Chem.1999,64,3885.doi:10.1021/jo982135h
(10) Fagnoni,M.;Mella,M.;Albini,A.Org.Lett.1999,1,1299.doi:10.1021/ol990982g
(11) Mukhopadhyay,S.;Rothenberg,G.;Gitis,D.;Sasson,Y.J.Org.Chem.2000,65,3107.doi:10.1021/jo991868e
(12)Inoue,A.;Kitagawa,K.;Shinokubo,H.;Oshima,K.Tetrahedron2000,56,9601.doi:10.1016/S0040-4020(00)00929-7
(13) Hassan,J.;Sévignon,M.;Gozzi,C.;Schulz,E.;Lemaire,M.Chem.Rev.2002,102,1359.doi:10.1021/cr000664r
(14)Wang,L.;Zhang,Y.;Liu,L.;Wang,Y.J.Org.Chem.2006,71,1284.doi:10.1021/jo052300a
(15) Dankwardt,J.W.Angew.Chem.Int.Edit.2004,116,2482.
(16)Dankwardt,J.W.J.Organomet.Chem.2005,690,932.doi:10.1016/j.jorganchem.2004.10.037
(17) Catellani,M.;Motti,E.;Della Ca,N.;Ferraccioli,R.Eur.J.Org.Chem.2007,2007,4153.
(18)Billingsley,K.L.;Barder,T.E.;Buchwald,S.L.Angew.Chem.Int.Edit.2007,119,5455.
(19)Zhou,Z.;Liu,M.;Wu,X.;Yu,H.;Xu,G.;Xie,Y.Appl.Organomet.Chem.2013,27,562.
(20) Breitenfeld,J.;Vechorkin,O.;Corminboeuf,C.;Scopelliti,R.;Hu,X.Organometallics2010,29,3686.doi:10.1021/om1007506
(21) Jana,R.;Pathak,T.P.;Sigman,M.S.Chem.Rev.2011,111,1417.doi:10.1021/cr100327p
(22)Amatore,M.;Gosmini,C.Angew.Chem.Int.Edit.2008,120,2119.
(23)Qian,Q.;Zang,Z.;Wang,S.;Chen,Y.;Lin,K.;Gong,H.Synlett2013,24,619.doi:10.1055/s-00000083
(24) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.et al.Gaussian 03,Revision 01;Gaussian Inc.;Wallingford,CT,2004.
(25) Becke,A.D.Phys.Rev.A1988,38,3098.doi:10.1103/PhysRevA.38.3098
(26) Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/1.464913
(27) Lee,C.T.;Yang,W.T.;Parr,R.G.Phys.Rev.B1988,37,785.doi:10.1103/PhysRevB.37.785
(28) Stephens,P.J.;Devlin,F.J.;Chabalowski,C.F.;Frisch,M.J.J.Phys.Chem.1994,98,11623.doi:10.1021/j100096a001
(29) Krishnan,R.;Binkley,J.S.;Seeger,R.;Pople,J.A.J.Chem.Phys.1980,72,650.doi:10.1063/1.438955
(30)McLean,A.D.;Chandler,G.S.J.Chem.Phys.1980,72,5639.doi:10.1063/1.438980
(31)Andrae,D.;Haussermann,U.;Dolg,M.;Stoll,H.;Preuss,H.Theor.Chim.Acta1990,77,123.doi:10.1007/BF01114537
(32)Cossi,M.;Rega,N.;Scalmani,G.;Barone,V.J.Comput.Chem.2003,24,669.doi:10.1002/jcc.10189
(33)Lin,B.L.;Liu,L.;Fu,Y.;Luo,S.W.;Chen,Q.;Guo,Q.X.Organometallics2004,23,2114.doi:10.1021/om034067h
(34) Liu,Y.;Liu,J.W.;Yang,X.Z.Acta Phys.-Chim.Sin.2002,18,1068.[劉 躍,劉佳雯,楊小震.物理化學(xué)學(xué)報(bào),2002,18,1068.]doi:10.3866/PKU.WHXB20021203
(35) Li,Z.;Jiang,Y.Y.;Fu,Y.Chem.Eur.J.2012,18,4345.doi:10.1002/chem.v18.14
(36) Lin,X.;Phillips,D.L.J.Org.Chem.2008,73,3680.doi:10.1021/jo702497p
(37) Joshi-Pangu,A.;Ganesh,M.;Biscoe,M.R.Org.Lett.2011,13,1218.doi:10.1021/ol200098d
(38)Tsou,T.T.;Kochi,J.K.J.Am.Chem.Soc.1979,101,6319.doi:10.1021/ja00515a028
(39)Bakac,A.;Espenson,J.H.J.Am.Chem.Soc.1986,108,719.doi:10.1021/ja00264a024
(40) Besora,M.;Carreón-Macedo,J.L.;Cimas,á.;Harvey,J.N.Adv.Inorg.Chem.2009,61,573.doi:10.1016/S0898-8838(09)00210-4
(41) Phapale,V.B.;Guisán-Ceinos,M.;Bu?uel,E.;Cárdenas,D.J.Chem.Eur.J.2009,15,12681.doi:10.1002/chem.v15:46
(42)Moncomble,A.;Le Floch,P.;Gosmini,C.Chem.Eur.J.2009,15,4770.doi:10.1002/chem.v15:19
(43) Li,Z.;Zhang,S.L.;Fu,Y.;Guo,Q.X.;Liu,L.J.Am.Chem.Soc.2009,131,8815.doi:10.1021/ja810157e
(44)Czaplik,W.M.;Mayer,M.;Jacobi von Wangelin,A.Angew.Chem.Int.Edit.2009,48,607.doi:10.1002/anie.v48:3
(45)Amatore,M.;Gosmini,C.Chem.Commun.2008,5019.
(46) Krasovskiy,A.;Duplais,C.;Lipshutz,B.H.J.Am.Chem.Soc.2009,131,15592.doi:10.1021/ja906803t
(47) Everson,D.A.;Jones,B.A.;Weix,D.J.J.Am.Chem.Soc.2012,134,6146.doi:10.1021/ja301769r
(48) Jiang,F.;Ren,Q.J.Organomet.Chem.2014,757,72.doi:10.1016/j.jorganchem.2013.12.047
猜你喜歡
無(wú)機(jī)化學(xué)學(xué)報(bào)(2023年12期)2023-12-21 01:01:50
昆明理工大學(xué)學(xué)報(bào)·社科版(2023年2期)2023-05-08 07:57:26
昆明理工大學(xué)學(xué)報(bào)·社科版(2023年2期)2023-05-08 07:57:16
環(huán)境衛(wèi)生工程(2021年5期)2021-11-20 05:45:30
發(fā)光學(xué)報(bào)(2021年6期)2021-06-16 13:39:32
首都師范大學(xué)學(xué)報(bào)(自然科學(xué)版)(2019年5期)2019-10-16 11:03:44
無(wú)機(jī)化學(xué)學(xué)報(bào)(2018年12期)2018-12-10 06:49:10
上海計(jì)量測(cè)試(2018年3期)2018-07-09 11:49:36
———理學(xué)院
西安航空學(xué)院學(xué)報(bào)(2018年1期)2018-02-05 09:49:15
物理化學(xué)學(xué)報(bào)(2013年7期)2013-09-21 09:00:42

- 物理化學(xué)學(xué)報(bào)的其它文章
- 浸漬法制備的Pd-MnO x/γ-Al2O3催化劑及不同載體對(duì)地表O3降解的影響
- MnO2/NiCo2O4的靜電自組裝合成及其電化學(xué)性能
- 碳摻雜α-S8的光學(xué)性質(zhì)和彈性性質(zhì)的第一性原理計(jì)算
- C2(a3Пu)自由基與含硫小分子反應(yīng)的溫度效應(yīng)
- Pt-Ni Catalyst Supported on CMK-5 for the Electrochemical Oxidation of Methanol
- Controlled Synthesis of Mesoporous MnO2Nanospindles