
Simulation of Liquid Argon Flow along a Nanochannel: Effect of Applied Force*
2009-05-15 06:17:52YINChunYangandElHarbawiMohanad
YIN Chun-Yang** and El-Harbawi Mohanad
?
Simulation of Liquid Argon Flow along a Nanochannel: Effect of Applied Force*
YIN Chun-Yang1,** and El-Harbawi Mohanad2
1Faculty of Chemical Engineering, Universiti Teknologi MARA, Shah Alam, 40450, Selangor, Malaysia2Chemical Engineering Department, Universiti Teknologi PETRONAS, Bandar Seri Iskandar, 31750 Tronoh, Perak, Malaysia
Liquid argon flow along a nanochannel is studied using molecular dynamics (MD) simulation in this work. Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) is used as the MD simulator. The effects of reduced forces at 0.5, 1.0 and 2.0 on argon flow on system energy in the form of system potential energy, pressure and velocity profile are described. Output in the form of three-dimensional visualization of the system at steady-state condition using Visual Molecular Dynamics (VMD) is provided to describe the dynamics of the argon atoms. The equilibrium state is reached after 16000 time steps. The effects on system energy, pressure and velocity profile due to reduced force of 2.0 (F2) are clearly distinguishable from the other two lower forces where sufficiently high net force along the direction of the nanochannel for F2 renders the attractive and repulsive forces between the argon atoms virtually no-existent. A reduced force of 0.5 (F0.5) provides liquid argon flow that approaches Poiseuille (laminar) flow as clearly shown by the-shaped average velocity profile. The extension of the present MD model to a more practical application affords scientists and engineers a good option for simulation of other nanofluidic dynamics processes.
molecular dynamics, large-scale atomic/molecular massively parallel simulator, visual molecular dynamics, nanofluidics, argon
1 INTRODUCTION
Molecular dynamics (MD) is a simulation method which utilizes Newton’s second law of motion to simulate the inter-atomic behavior of individual atomsstatistical mechanics. With the creation of computers, MD simulations have been made considerably easier due to their fast and powerful computational capabilities. In recent times, engineers have begun to recognize the value of MD simulations which can be applied to various engineering simulations. It is essential for engineers to fully capitalize on the capability of MD simulations which can generate primary data to accurately predict macroscopic properties.
A complete understanding of the properties of fluid interfaces is essential from the technical and theoretical point of view [1]. The surface properties of fluids in pores play a very important role in many fields and influence the transport properties in many multiphase systems [2]. As such, computer simulation such as MD can afford scientists and engineers ways to determine these surface properties without the need to conduct laborious experimental works. Such simulations can assist in describing nanofluidic flows which normally cannot be described accuratelymacroscopic principles. There have been numerous studies dedicated to simulation of argon flow along nanochannels [2-4]. There are, however, few studies on such flows subjected to different applied forces. This is important in MD simulations as inappropriately specified applied force will either hinder effective measurement of fluxes or render non-linearity of system response [3]. In a sense, an applied constant force along a nanochannel has the same influence as permitting gravity to initiate the nanofluidic flow. Xu and Zhou [4] commented that at very small non-dimensional gravity force (or reduced force), for most cases less than 0.05, the gravity force has no contribution to the mean axial velocity. In this case, the liquid molecules oscillate randomly in three coordinates (,and) due to the intermolecular force with zero mean velocities. As such, it can be seen from previous related studies that applied reduced forces are usually stipulated at magnitudes higher than 0.05.
The objective of this paper is to simulate liquid argon flow in a slit-like nanochannel using MD simulation. The reason for selecting argon as the fluid for simulation is that the atoms have well-established atomic potential interaction and it serves as a reference point for more complex fluid simulations. The effects of applied force on the argon flow on system energy in the form of potential energy, pressure and velocity profile are described. An output in the form of 3-D visualization of the system is also provided to fully describe the dynamics of the argon atoms. Usage of specialized open-source MD simulation software for this study represents a novel aspect of the study in addition to reporting new simulation results.
2 SIMULATION
In this study, Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) [5], which is an open-source software written in C++ and developed at Sandia National Labs is used for this MD simulation. LAMMPS is selected for this study due to its simplicity and ease of operation. Dimensionless physical quantities (Lennard-Jones reduced units) are used for convenience as well as to facilitate scale-up of the system. A simple fluid of argon using Lennard-Jones (12-6) potential as expressed in single atom situation is considered:
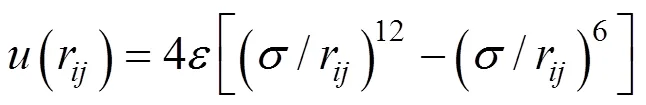
where(r) is the interaction potential between two particles,the depth of the potential well that governs the strength of interaction and σ the finite distance where the interparticle potential is zero andris the distance between the two particles [6]. Table 1 lists the units used in our argon MD simulation [7] whereis the Boltzmann constant. Hereafter, all quantities will be given in terms of Lennard-Jones reduced units.
The simulation region is a cuboid with unit lengths of 10, 4 and 3 (molecular units) along-,- and-directions respectively as required in LAMMPS input file. Periodic boundary conditions are applied along- and-directions while-direction is applied with a non-periodic and shrink-wrapped boundary. The shrink-wrapped boundary is taken to mean that the position of the face is set so as to encompass the atoms in that dimension, no matter how far they move [8]. In other words, when a particle enters or leaves the simulation region, an image particle leaves or enters this region, such that the number of particles from the simulation region is always conserved [7]. In our simulation, we apply a constant force to each particle along the nanochannel (-direction). The wall and fluid atoms have the same Lennard-Jones parameters and the same mass [3] and the wall atoms are stationary. The fluid atoms are arranged in face-cubic-center (FCC) lattice and each atom has a mass of 1.0. There are 60 argon atoms in each single layer wall and 420 fluid atoms (Fig. 1). The cut-off distance is specified to be 21/6(Weeks-Chandler-Andersen potential) [3]. It should be noted that any argon atom within the simulation region has interaction with neighboring fluid and wall atoms. The specific applied force on fluid atoms is specified to be 0.5, 1.0 or 2.0 in the-direction with constant temperature of 1.0. The simulation runs consist of 200000 time steps with a reduced time step of 0.001. Constantintegration (ensemble) is performed to update position and velocity for atoms in the group each time step whereis volume whileis energy.
3 RESULTS AND DISCUSSION
3.1 System pressure
A typical LAMMPS output file consists of the coordinates of every atom printed in everytime steps, whereis a number specified in the input script in which VMD reads the output file and shows a visual representation of how the simulation progresses [9]. During the simulation run, thermodynamic data is provided every time steps. After the end of the run, LAMMPS prints the final thermodynamic state and a total run time for the simulation. For convenience and brevity, hereafter F0.5, F1 and F2 shall represent the system at applied force of 0.5, 1.0 and 2.0 respectively. Fig. 2 shows the reduced pressure of the system throughout the simulation run. Due to addition of force at the beginning of the run, the pressure of the system drastically increases from 0.6 to approximately 4.0 at timesteps of 1000 (for all forces) before gradually reduces to 3.4 (F0.5), 2.2 (F1) and 0.61 (F2) at time step 16000. The effect of applied force at the initial stage causes random displacement of argon atoms. The atoms gradually pick up momentum due to the force action and travel along the nanochannel, thus gradually reduces the system pressure. It is interesting to note that after the system pressure reaches relative “equilibrium” at time step 16000, pressure of F2 returns to its initial value while F0.5 and F1 have significantly higher system pressures. In addition, the system pressures for F0.5 and F1 fluctuate rather widely after 16000 time steps while pressure of F2 remains constant. In a qualitative sense, we suggest that these observed phenomena are attributed to sufficiently high net force in the-direction for F2 which renders the attractive and repulsive forces between the argon atoms virtually non-existent. Since these attractive and repulsive forces occur in all directions, it can be seen that this causes both reduction and increase in system pressures at varying magnitudes for F0.5 and F1. This is further elaborated in subsequent sections.
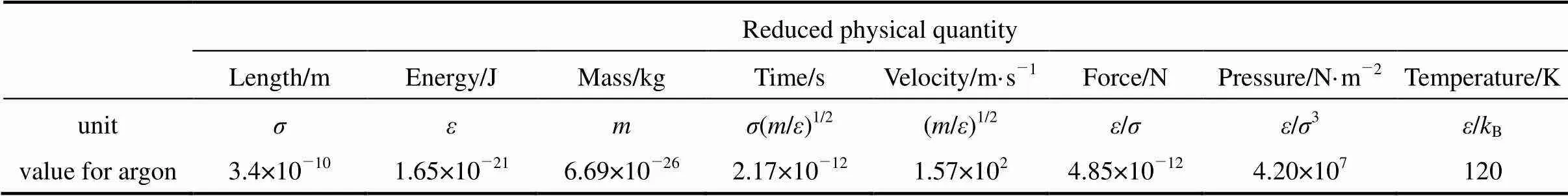
Table 1 Units used in MD simulation of particles interacting by the Lennard-Jones potential [7]
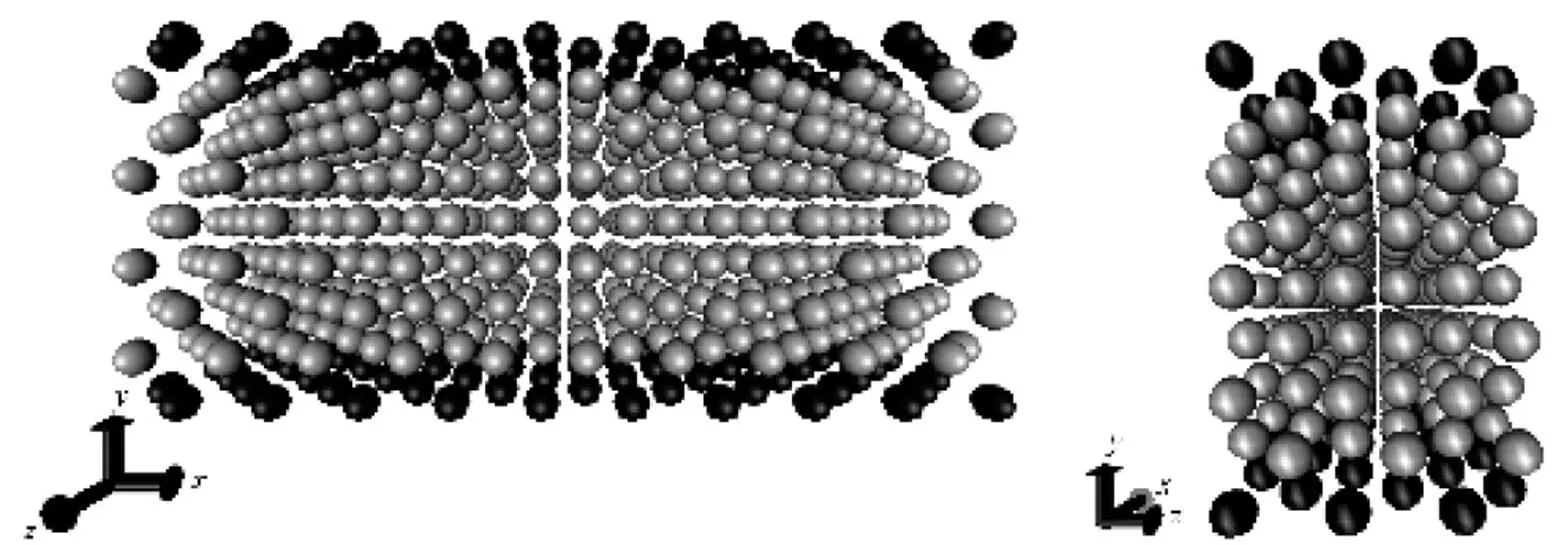
Figure 1 Initial ordered FCC configuration of the argon atoms in a simulation cuboid cell observed from two different angles
(The wall atoms are in black while the fluid atoms are in grey-white)
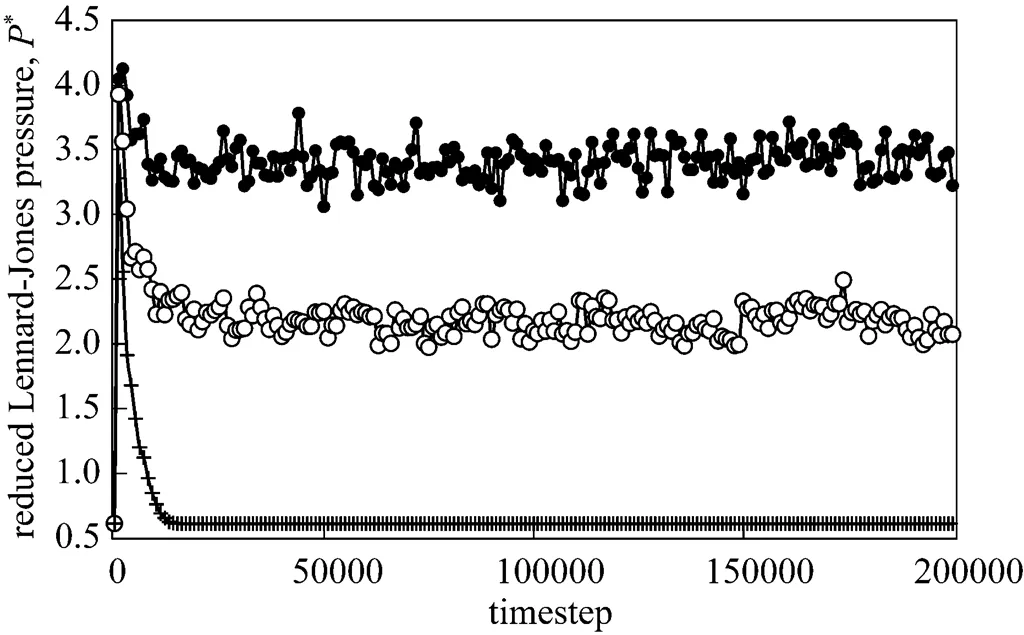
Figure 2 Reduced pressure of system throughout the MD simulation run
force:●?0.5;○?1.0; + 2.0
3.2 Potential energy
Figure 3 shows the evolution of potential energy throughout the simulation run. The curve trend of the potential energy is somewhat analogous to the pressure curve in which an equilibrium state is achieved for F2. As can be described from Eq. (1), there is therefore, a change in distance between two or more particles, resulting in fluctuations in van der Waals attraction and repelling forces. Due to the type of system,, which has been used during this simulation, the total energy does not change as soon as an equilibrium state is reached [10]. This state is obtained after 16000 time steps. This indicates “convergence” of the computed values is achieved and implies that for this system, a run length of more than 16000 must be conducted in order for a reliable ‘steady-state’ result to be obtained. For F2, at time steps of 4000, the variation of potential energy (compared to the start of the simulation) is the highest and the atoms are randomly displaced from their original positions due to force acting along-direction. At time steps between 4000 and 16000, the atoms slowly pick up momentum and move along the nanochannel. At this time, the atoms are still attracting and repelling one another as they still contain excess potential energy. From time step 16000 to the end of simulation, there is no excess potential energy and the atoms freely move along the nanochannel with no significant displacement along-axis.

Figure 3 Evolution of potential energy throughout the MD simulation run
force: ○?0.5;●?1.0; △?2.0
3.3 Visualization

Figure 4 VMD snapshots from two different angles at timesteps of 200000 for different forces (The wall atoms are in black
while the fluid atoms are in grey-white)
3.4 Velocity profile
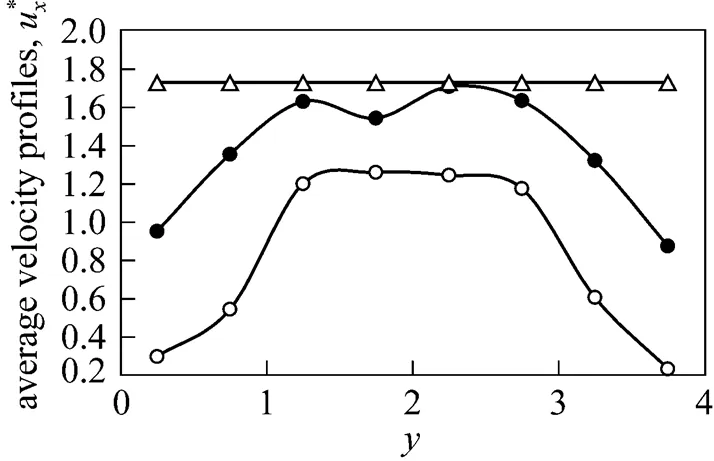
Figure 5 Average velocity profiles
force:○?0.5;●?1.0;△?2.0
As expected, increase in applied force results in increased average velocity of the fluid. Again, outcome from F2 deviates from F0.5 and F1 where a single uniform average velocity of 1.73 is observed across the width of the channel. This observation is in agreement with the abovementioned results where sufficiently high net force in the-direction for F2 renders the interactive forces between the argon atoms insignificant. Our observation is consistent with a condition elucidated by Xu and Zhou [4], where an applied force of 5 causes a uniform mean velocity profile of argon atoms across two platinum walls, with a linear increase with time at a constant acceleration of the non-dimensional gravity force. This condition is known as the “force-domain” condition while the flow for F0.5 and F1 exist in “coupling” condition where flow is governed by not only the applied gravity force, but also the intermolecular force due to the Lennard- ones potential [4].
The velocity profile curve for F1 can be correlated with relative accuracythe following quadratic function meant for “coupling” condition [4]:
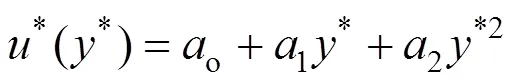
4 CONCLUSIONS
Using MD simulation of a simple argon flow along a nanochannel, the displacement of fluidic atoms due to applied forces as well as interatomic forces has been demonstrated. The equilibrium state is reached after 16000 time steps. The effects on system energy, pressure and velocity profile due to applied force of 2 are clearly distinguishable from the other two lower forces where sufficiently high net force in the-direction for F2 renders the attractive and repulsive forces between the argon atoms virtually non-existent. The visual dynamics projection afforded by VMD shows this effect distinctly where the majority of the F2 fluid atoms appear to move along-direction at a regular and faster pace, seemingly receiving marginal interference caused by neighbouring forces. The average velocity profiles for F0.5 and F1 exhibit a characteristic-shape indicating interaction of fluid atoms with wall atoms while for F2, a single uniform average velocity of 1.73 is observed across the width of the channel. F0.5 provides liquid argon flow that approaches Poiseuille flow. The extension of the current MD model to a more practical application affords scientists and engineers a good optionfor simulation of other nanofluidic dynamics processes.
1 Meyer, M., Mareschal, M., Hayoun, M., “Computer modeling of a liquid-liquid interface”,..., 89, 1067-1073 (1988).
2 Zhang, H., Zhang, B.J., Lu, J., Liang, S., “Molecular dynamics simulations on the adsorption and surface phenomenon of simple fluid in porous media”,..., 366, 24-27 (2002).
3 Travis, K.P., Gubbins, K.E., “Poiseuille flow of Lennard-Jones fluids in narrow slit pores”,..., 112, 1984-1994 (2000).
4 Xu, J.L., Zhou, Z.Q., “Molecular dynamics simulation of liquid argon flow at platinum surfaces”,, 40, 859-869 (2004).
5 Plimpton, S. J., “Fast parallel algorithms for short-range molecular dynamics”,..., 117, 1-19 (1995).
6 Lennard-Jones, J.E., “Cohesion”,..., 43, 461-482 (1931).
7 Beu, T.A., Molecular Dynamics Simulations, University Babes-Bolyai, Cluj-Napoca, Romania (2002).
8 Plimpton, S.J., Crozier, P., Thompson, A., LAMMPS User Manual, Sandia National Laboratories, USA (2003).
9 Fried, J., “Numerical simulation of viscous flow: A study of molecular dynamics and computational fluid dynamics”, M.Sc. Thesis, Virginia Polytechnic Institute and State Univ., USA (2007).
10 Jegan, P., “Further investigation of molecular dynamics simulations for argon adsorption on single-walled carbon nanotube bundles”, M.Sc. Thesis, Cranfield Univ., United Kingdom (2007).
11 Humphrey, W., Dalke, A., Schulten, K., “VMD-visual molecular dynamics”,..., 4, 33-38 (1996).
12 Ziarani, A.S., Mohamad, A.A., “A molecular dynamics study of perturbed Poiseuille flow in a nanochannel”,., 2, 12-20 (2006).
13 Song, X., Chen, J.K., “A comparative study on Poiseuille flow of simple fluids through cylindrical and slit-like nanochannels”,.., 51, 1770-1779 (2008).
2009-03-23,
2009-09-03.
the Academy of Sciences, Malaysia and Ministry of Science and Technology & Innovation.
** To whom correspondence should be addressed. E-mail: yinyang@salam.uitm.edu.my
 Chinese Journal of Chemical Engineering2009年5期
Chinese Journal of Chemical Engineering2009年5期
- Chinese Journal of Chemical Engineering的其它文章
- Effect of Relative Humidity on Catalytic Combustion of Toluene over Copper Based Catalysts with Different Supports*
- Prediction of Critical Endpoints Based on the PR Equation of State*
- Kinetic Studies on Wheat Straw Hydrolysis to Levulinic Acid*
- Calculation of Transport Properties of CF4+Noble Gas Mixtures
- Synthesis of 4,4′-MDC in the Presence of Sulfonic Acid-functionalized Ionic Liquids*
- A New Study on Combustion Behavior of Pine Sawdust Characterized by the Weibull Distribution*