
Targeted next-generation sequencing identifies a novel nonsense mutation in ANK1 for hereditary spherocytosis: A case report
2022-06-22 08:49:16PanFuYangYangJiaoKaiChenJingBoShaoXueLianLiaoJingWeiYangShaYiJiang
World Journal of Clinical Cases 2022年15期
INTRODUCTION
Hereditary spherocytosis (MIM: 182900) is the third most common hemolytic disease after glucose-6-phosphate dehydrogenase deficiency and ABO hemolytic disease[1]. HS is a common, inherited, red cell membrane disorder with an incidence of 1/2000 in Northern Europe and Northern America[2], whereas in China it is considered less common and affects approximately 1.27 cases per 100000 people in males and 1.49 cases per 100000 people in females[3]; however, these discrepancies may reflect a considerable number of undiagnosed, asymptomatic cases. HS is fundamentally characterized by a mechanical abnormality of erythrocytes, along with osmotic changes, and can result in kernicterus in newborn babies. The main biochemical defects of HS lie in the proteins of the erythrocyte membrane, including ankyrin, band 3, α-spectrin, β-spectrin, and protein 4.2, which are encoded by the
and
genes, respectively[4]. Approximately 50% of patients with HS present with anemia and 10 to 15% with jaundice or splenomegaly[5]. The symptoms of HS can vary widely from asymptomatic patients to those that require blood transfusions or even life-threatening anemia. An initial symptom of HS in neonates may be hyperbilirubinemia that is unrelated to blood incompatibility.Here, we present a HS case with a novel mutation of
present within a Chinese family.
CASE PRESENTATION
Chief complaints
A pale complexion for more than 2 mo.
On the other hand, without encouragement talented students may never be motivated to learn, develop skills, or reach their full potential. For example, at the same high school, there was a teacher whose Spanish language classes I attended but from whom I, unfortunately learned very little simply because of the woman’s cold sarcastically8 critical attitude. She seemed to know nothing about encouraging students, and she was gifted speaking contemptuously of those of us who weren’t learning fast enough. Her negativism drove me away. Partly because of this teacher’s negative influence, I am not fluent in Spanish today.
History of present illness
A female neonate displayed splenomegaly and moderate anemia with a Hb level of 60 g/L at examination 1 mo after birth. The girl was then taken to Shanghai Children’s Hospital for further treatment 3 mo later.
History of past illness
The child was pale at the age of 1 mo. Anemia was detected in the blood after a routine examination at the local hospital, but no treatment was given.
Personal and family history
G1P1, gestational age 39 wk, birth weight 3110 g, she was the first child of the family, and her father currently has mild splenomegaly but no anemia and no history of blood transfusion.
When I was growing up, my father always stopped what he was doing and listened while I d breathlessly fill him in on my day. For him, no subject was off-limits. When I was a lanky1 and awkward 13, Dad coached me on how to stand and walk like a lady. At 17 and madly in love, I sought his advice on pursuing a new student at school. “Keep the conversation neutral,” he counseled. “And ask him about his car.”
Physical examination
The sea was quite black and thick, and it was breaking high on the beach; the foam18 was flying about, and the wind was blowing; everything looked bleak19
Laboratory examinations
Blood tests showed moderate anemia, reticulocytosis, and hyperbilirubinemia that was mainly unconjugated bilirubin (Table 1). An osmotic fragility test showed that a concentration of NaCl solution of 0.52% (ref: 0.38%-0.41%) initiated hemolysis and at a concentration of 0.44% (ref: 0.30%-0.34%) there was complete hemolysis; indicative of increased erythrocyte osmotic fragility. The activity of glucose-6-phosphate dehydrogenase (G-6-PD) was normal and a Coomb’s test was negative. Hemoglobin electrophoresis, and α- and β-globin genetic analysis excluded α- and β-thalassemia. A significant increase of spherocytes after a peripheral blood smear was absent.

Imaging examinations
A chest X-ray showed no active lesions in the lungs. Echocardiography showed an atrial septal defect(1.2 mm) and the oval foramen was not closed.
Genetic testing
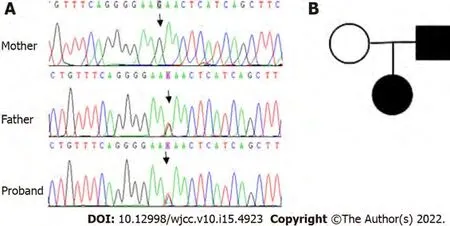
To identify the cause of the unexplained hemolysis, genetic analysis by next-generation sequencing(NGS) was performed. DNA was extracted from peripheral blood collected before transfusion, and genetic analysis was conducted on 700 genes associated with hematological diseases. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit according to the manufacturer’s instructions. The DNA sample was quantified by Qubit dsDNA BR Assay kit. All libraries were prepared using the KAPA HTP Library Preparation Kit according to the manufacturer’s instructions. Fragmented DNA was repaired, 3’ dA-tailed, ligated with Illumina adapters, size selected, amplified, and assessed using the Agilent 2100 Bioanalyzer. The captured DNA library was finally amplified and sequenced on an Illumina Novoseq 6000 sequencer for paired reads at 150 bp. The original data were converted from bcl files to fastq format files by Illumina CASAVA1.8 (Illumina, San Diego, CA, United States), and reads were compared to the GRCh37/hg19 human genome reference using BWA, samtools, picard, and GATK to remove repeated sequences and identify genetic variants. All of the identified variants were evaluated by browsing through databases, including NCBI dbSNP, OMIM, HGMD and NCBI ClinVar.The putative effects on the ANK1 protein of all the identified variants were explored using a variety of prediction algorithms, including PolyPhen2, SIFT and Mutation Taster. Finally, a heterozygous mutation in
(exon23:c.G2467T:p.E823X) was detected in the girl and her father, whereas her mother was wild type (Figure 1).
Fu P and Jiang SY analyzed the case and wrote and revised the manuscript; Jiao YY, Liao XL,and Yang JW helped collect the original data; All authors participated in the treatment of the patient.
FINAL DIAGNOSIS
Hereditary spherocytosis.
TREATMENT
She received suspension RBCs transfusion during her hospitalization.
OUTCOME AND FOLLOW-UP
To date, and 4 mo after her last blood transfusion, the child’s hemoglobin levels have fluctuated between 90 g/L and 100 g/L and a further blood transfusion was not required.
DISCUSSION
Mutations within the
gene are the most common cause of HS (up to 50% of cases), followed by mutations in the spectrin gene (
, up to 20%;
, up to 5%),
(up to 15%), and
(up to 10%)[6]. It has been reported that heterozygous
mutations account for 52% of all Korean HS patients and approximately 31% of all Japanese HS patients[7,8]. The
gene is located at 8p11.21[9], contains 42 exons, and with a cDNA of 8300 bp in length. The
gene encodes a critical component of the erythrocyte membrane skeleton, the ankyrin 1 protein, which is composed of 1881 amino acids and possesses three main structural domains[10]. Each erythrocyte contains approximately 1.24 × 10
ankyrins[8].
mutations are primarily frameshift, nonsense, and splice site mutations[7,11], and it is frequently mutated
. Most of the mutations occur in the coding sequence of genes and can result in functional deficiencies of the protein. Autosomal recessive inheritance has occasionally been recorded in a few patients[12]. For recessive HS, the common mutational types are missense mutations and with a mutation in the putative ankyrin-1 promoter[13].
A further physical examination confirmed pallor and splenomegaly.
“The Power of twelve men!” said the Finland woman; “that would be of very little use.” But she went to a shelf and took down and unrolled a large skin, on which were inscribed6 wonderful characters, and she read till the perspiration7 ran down from her forehead. But the reindeer begged so hard for little Gerda, and Gerda looked at the Finland woman with such beseeching8 tearful eyes, that her own eyes began to twinkle again; so she drew the reindeer into a corner, and whispered to him while she laid a fresh piece of ice on his head, “Little Kay is really with the Snow Queen, but he finds everything there so much to his taste and his liking9, that he believes it is the finest place in the world; but this is because he has a piece of broken glass in his heart, and a little piece of glass in his eye. These must be taken out, or he will never be a human being again, and the Snow Queen will retain her power over him.”
According to the HGMD, ExAC, gnomAD, and 1000 genomes databases, the detected mutation(ANK1:exon23:c.G2467T:p.E823X) in this Chinese family led to premature termination of the protein,thereby forming a truncated protein without normal function. Since the mutation is novel, the clinical manifestations associated with it are unclear. At the time of diagnosis in early infancy, the proband had splenomegaly and moderate anemia that required a blood transfusion. However, at present, the degree of anemia is reduced, and the patient is not dependent upon a blood transfusion. Similarly, her father displayed a history of anemia in his childhood, but his current symptoms are now only mild. Therefore,it is speculated that this mutation leads to relatively serious anemia after birth, but that anemia improves with age. The diagnostics for HS in this family were based upon laboratory findings and clinical examinations, as well as a positive family history, and we found a novel nonsense mutation of
, detected by targeted next generation sequencing and identified by Sanger sequencing in this study.
They drank their coffee and had a chat together, and then AnneLisbeth went away towards the little town where she was to meet thecarrier, who was to drive her back to her own home. But when shecame to him she found that he would not be ready to start till theevening of the next day. Then she began to think of the expense, andwhat the distance would be to walk. She remembered that the route by the sea-shore was two miles shorter than by the high road; and asthe weather was clear, and there would be moonlight, she determined to make her way on foot, and to start at once, that she might reachhome the next day.
A novel
mutation considered causative of HS was identified in a Chinese family and its clinical features were elucidated and documented. This novel study expands the current spectrum of
mutations. Moreover, the analysis of pathogenic gene mutations
NGS and Sanger sequencing can provide a powerful tool to support HS diagnosis and the associated genetic consultation of HS patients.However, the pathogenic mechanism of the
mutation is unclear and needs to be explored further to help with the diagnosis of HS.
Little One-eye sat down, and as she was very much tired by the long walk to which she was not used, and by the hot day, and as Little Two-eyes went on singing
When compared directly with Sanger sequencing, NGS has higher diagnostic efficiency for suspected red blood cell membrane disorders in patients[17,18]. In addition to helping to confirm a diagnosis,genetic technology also helps to assess the risk of HS for descendants of a family and provide information for potential genetic counselling and future research and understanding of HS.
Regarding treatment, splenectomy, as the standard surgical treatment in moderate and severe patients with HS[19], is efficient but does have drawbacks, of which the most recognized is a risk of infection. In addition, the heterogeneity of clinical manifestations requires close observation of the disease progression to appropriately determine the timing of surgery. Before undertaking a splenectomy, the diagnosis should be confirmed by clinical data and a genetic examination.
CONCLUSION
In general, children with Coomb’s negative hemolytic disease can be diagnosed as HS according to a family history of HS, positive osmotic fragility test, spherocytosis on a blood smear, and the exclusion of possible causes of secondary spherocytosis. Yet, 20% of HS patients do not display typical spherocytosis;hence, for such patients without spherocytosis, genetic testing is very important to make a diagnosis of HS. It has been reported that splenectomy was required for a girl with highly suspected HS and with an anemia recovery, and then decades later, her son was subsequently diagnosed with HS by genetic testing[12]. With the rapid development and wide clinical application of gene technologies, a growing number of HS cases have been identified by genetic tests, especially asymptomatic patients. This has led to the identification of several new mutants in HS genes in our center through this gene testing[14].Guideline recommendations are that further molecular testing is not a requirement when a family history, clinical manifestations, and laboratory tests all support the diagnosis of HS[15]. However, in view of the fact that there are no hotspot mutations in HS and most mutations are sporadic and specific to individual patients or their families[16], it is necessary to detect and characterize gene mutations in patients with atypical clinical presentations.
FOOTNOTES
Many analysts82 believe animal bridegroom tales are intended to alleviate83 a maiden s fears of the marriage bed. While her husband may appear to be a beast before their marriage, she will learn that he is simply a caring man once the marriage is consummated84.Return to place in story.
The complexity of gene mutations and gene regulation may contribute to the heterogeneity of clinical manifestations. Patients with
gene mutations tend to be more anemic and with a higher level of reticulocytosis than those without[8]
However, even if the mutation site is the same, disease severity can greatly differ[14], and furthermore, for the same patient, the degree of anemia at any given period can also vary extensively. We report here a new mutation in a family with HS in which the child showed initial transfusion dependence during the early infantile period; however, the anemia improved with age.
the Natural Science Foundation of Shanghai Science Committee, No. 18ZR1431200; Research Foundation of Shanghai Municipal Health Commission, No. 20194Y0112; and Clinical Research Plan of SHDC, No.SHDC2020CR4089.
He had the miller s daughter put into another room full of straw, much bigger than the first, and bade her, if she valued her life, spin it all into gold before the following morning
The parents of the patient consented to the publication of the case and any accompanying images with written consent.
The authors declare that they have no competing interests.
The authors have read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
China
Jing-Bo Shao 0000-0002-1451-8033; Sha-Yi Jiang 0000-0002-6734-9122.
Ma YJ
Filipodia
Ma YJ
1 Christensen RD, Yaish HM, Gallagher PG. A pediatrician's practical guide to diagnosing and treating hereditary spherocytosis in neonates.
2015; 135: 1107-1114 [PMID: 26009624 DOI: 10.1542/peds.2014-3516]
2 Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders.
2013; 27: 167-178 [PMID: 23664421 DOI: 10.1016/j.blre.2013.04.003]
3 Wang C, Cui Y, Li Y, Liu X, Han J. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model.
2015; 4: 76-81 [PMID: 25984425 DOI: 10.5582/irdr.2015.01002]
4 Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis.
2008; 372: 1411-1426 [PMID: 18940465 DOI:10.1016/S0140-6736(08)61588-3]
5 Krueger HC, Burgert EO Jr. Hereditary spherocytosis in 100 children.
1966; 41: 821-830 [PMID:5926777]
6 An X, Mohandas N. Disorders of red cell membrane.
2008; 141: 367-375 [PMID: 18341630 DOI:10.1111/j.1365-2141.2008.07091.x]
7 Park J, Jeong DC, Yoo J, Jang W, Chae H, Kim J, Kwon A, Choi H, Lee JW, Chung NG, Kim M, Kim Y. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis.
2016; 90: 69-78 [PMID: 26830532 DOI:10.1111/cge.12749]
8 Nakanishi H, Kanzaki A, Yawata A, Yamada O, Yawata Y. Ankyrin gene mutations in japanese patients with hereditary spherocytosis.
2001; 73: 54-63 [PMID: 11372755 DOI: 10.1007/BF02981903]
9 Lambert S, Yu H, Prchal JT, Lawler J, Ruff P, Speicher D, Cheung MC, Kan YW, Palek J. cDNA sequence for human erythrocyte ankyrin.
1990; 87: 1730-1734 [PMID: 1689849 DOI: 10.1073/pnas.87.5.1730]
10 Lux SE, John KM, Bennett V. Analysis of cDNA for human erythrocyte ankyrin indicates a repeated structure with homology to tissue-differentiation and cell-cycle control proteins.
1990; 344: 36-42 [PMID: 2137557 DOI:10.1038/344036a0]
11 Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer.
2004; 41: 118-141 [PMID: 15071790 DOI: 10.1053/j.seminhematol.2004.01.002]
12 Han JH, Kim S, Jang H, Kim SW, Lee MG, Koh H, Lee JH. Identification of a novel p.Q1772X ANK1 mutation in a Korean family with hereditary spherocytosis.
2015; 10: e0131251 [PMID: 26107955 DOI:10.1371/journal.pone.0131251]
13 Eber SW, Gonzalez JM, Lux ML, Scarpa AL, Tse WT, Dornwell M, Herbers J, Kugler W, Ozcan R, Pekrun A, Gallagher PG, Schr?ter W, Forget BG, Lux SE. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis.
1996; 13: 214-218 [PMID: 8640229 DOI: 10.1038/ng0696-214]
14 Hao L, Li S, Ma D, Chen S, Zhang B, Xiao D, Zhang J, Jiang N, Jiang S, Ma J. Two novel ANK1 loss-of-function mutations in Chinese families with hereditary spherocytosis.
2019; 23: 4454-4463 [PMID: 31016877 DOI:10.1111/jcmm.14343]
15 Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ; General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update.
2012; 156: 37-49 [PMID: 22055020 DOI: 10.1111/j.1365-2141.2011.08921.x]
16 He BJ, Liao L, Deng ZF, Tao YF, Xu YC, Lin FQ. Molecular Genetic Mechanisms of Hereditary Spherocytosis: Current Perspectives.
2018; 139: 60-66 [PMID: 29402830 DOI: 10.1159/000486229]
17 van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The Third Revolution in Sequencing Technology.
2018;34: 666-681 [PMID: 29941292 DOI: 10.1016/j.tig.2018.05.008]
18 van Vuren A, van der Zwaag B, Huisjes R, Lak N, Bierings M, Gerritsen E, van Beers E, Bartels M, van Wijk R. The Complexity of Genotype-Phenotype Correlations in Hereditary Spherocytosis: A Cohort of 95 Patients: Genotype-Phenotype Correlation in Hereditary Spherocytosis.
2019; 3: e276 [PMID: 31723846 DOI:10.1097/HS9.0000000000000276]
19 Manciu S, Matei E, Trandafir B. Hereditary Spherocytosis - Diagnosis, Surgical Treatment and Outcomes. A Literature Review.
2017; 112: 110-116 [PMID: 28463670 DOI: 10.21614/chirurgia.112.2.110]
 World Journal of Clinical Cases2022年15期
World Journal of Clinical Cases2022年15期
- World Journal of Clinical Cases的其它文章
- Diet and intestinal bacterial overgrowth: Is there evidence?
- Spontaneous liver rupture following SARS-CoV-2 infection in late pregnancy: A case report
- Metastasis of liver cancer to the thyroid after surgery: A case report
- Solitary primary pulmonary synovial sarcoma: A case report
- Knot impingement after arthroscopic rotator cuff repair mimicking infection: A case report
- Clear aligner treatment for a four-year-old patient with anterior crossbite and facial asymmetry: A case report