
High-quality chromosome-level genome assembly of redlip mullet (Planiliza haematocheila)
2021-02-10 13:07:24NaZhaoHaoBingGuoLeiJiaQiuXiaDengChunHuaZhuBoZhang
Zoological Research 2021年6期
Na Zhao, Hao-Bing Guo, Lei Jia, Qiu-Xia Deng,Chun-Hua Zhu, Bo Zhang,,*
1 Southern Marine Science and Engineering Guangdong Laboratory (Zhanjiang), Fisheries College, Guangdong Ocean University, Zhanjiang, Guangdong 524088, China
2 Tianjin Fisheries Research Institute, Tianjin 300200, China
3 Shanghai Ocean University, Shanghai 201306, China
4 BGI-Qingdao, BGI-Shenzhen, Qingdao, Shandong 266555,China
In the present study, we successfully assembled a high-quality genome ofPlanilizahaematocheila(redlip mullet) based on Oxford Nanopore long read, single-tube long fragment read(stLFR), and Hi-C chromatin interaction sequencing.The size of theP.haematocheilagenome was 652.91 Mb.More than 93.8% of BUSCO genes were detected, and the N50 lengths of contigs and scaffolds reached 7.21 Mb and 28.01 Mb,respectively, thus demonstrating outstanding genome completeness and sequence continuity.A total of 21 045 protein-coding genes were predicted in the assembled genome, and 99.77% of those genes were functionally annotated.Comparative genomic and phylogenetic analyses revealed the adaptability ofP.haematocheilato complex living environments at the genomic level, highlighting its broad adaptability and resistance to multiple stresses as an important economic fish.The high-quality reference chromosome-level genome ofP.haematocheilaprovides a powerful genomic resource for further systematic study of Mugilidae.
Planilizahaematocheila(FishBase ID: 13000), which belongs to Mugiliformes, Mugilidae, is an economically value fish.This species can survive under different salinities and water quality, and it shows strong adaptability to hypoxia compared to other aquaculture fish (Qi et al., 2016).Thus,P.haematocheilais an excellent model for studying fish adaptation to complex environments.However, despite its economic value, research onP.haematocheilais slow, with a focus on geographical (Durand & Borsa, 2015) and seasonal resource distribution (Shen et al., 2011), population dynamics(Pankov et al., 2009), nutritional supplementation (Zhang et al., 2013), and disease prevention (Qi et al., 2016).At present,our understanding of the in-depth mechanisms underlying its biological processes remains poor, which may be due to a lack of good genetic resources.Liyanage et al.(2019)previously established a draft genome ofP.haematocheilaat the contig level, however better-quality genomes are needed to meet the higher requirements of analysis.In the current study, we successfully assembled a high-quality genome ofP.haematocheilavia a combination of Oxford Nanopore long read, stLFR, and Hi-C chromatin interaction sequencing.Comparative genomic and phylogenetic analyses revealed the adaptability ofP.haematocheilato complex living environments at the genomic level, highlighting its broad adaptability and resistance to multiple stresses as an important economic fish.This high-quality reference chromosome-level genome ofP.haematocheilaprovides a powerful genomic resource for further systematic studies of Mugilidae.
Liver, blood, and muscle tissue samples from aP.haematocheilafemale were used for DNA extraction.Fresh samples were obtained from the Bohai Sea by the Tianjin Fisheries Research Institute, China.All sequencing libraries were established based on high-quality purified DNA.The chromosome-level genome was accomplished with a mixed assembly strategy.The stLFR data were first used to conduct genome k-mer analysis.Clean reads with duplications removed were filtered using SOAPnuke and then analyzed using Jellyfish v2.2.6 to obtain a histogram.GenomeScope v1.0.0 converted the histogram to the final visual result, as shown in Figure 1A.Oxford Nanopore sequencing data were employed to assemble adenovocontig-level genome using wtdbg2 (parameters: -p 0 -k 15 -AS 2 -s 0.05 -L 5 000, as suggested by the software when assembling a genome <1 G in size using nanopore/ont data).The output was polished with Pilon using stLFR data, as the quality values of single bases in these reads were far more precise than the Nanopore reads.Lastly, contigs were assembled into scaffolds by mapping Hi-C read pairs to the polished assembly with HiC-Pro, Juicer,and 3D-DNA.Additional details are provided in the Supplementary Materials and Methods.TheP.haematocheilachromosome-level genome statistics are shown in Table 1.The length of the 24 chromosomes obtained by Hi-C ranged from 32.04 Mb to 20.79 Mb, covering 99.31% of the genome(Fei et al., 1985).The heatmap generated by 3D-DNA is shown in Figure 1B, with the clear boundaries between different chromosomes indicating strong interactions inside each chromosome.
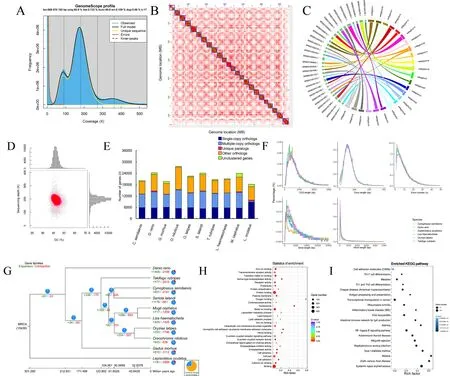
Figure 1 Statistics and data analysis of genome assembly of Planiliza haematocheila
We also compared the novel chromosome-level genome with the previously published contig-level genome (NCBI:GCA_005024645), which showed significant improvement in genome integrity.We not only assembled the genome into chromosome-level long scaffolds with a size closer to the kmer analysis result, but also virtually doubled the contig N50 value (as shown in Table 1).We selected the top 30 longest contigs of the previously published genome and mapped the sequences to ourP.haematocheilachromosome-level assembly.Lastz (http://www.bx.psu.edu/miller_lab/dist/README.lastz-1.02.00/README.lastz-1.02.00a.html) was employed to find the mapped fragments between the genomes, and the minimum block size of the mapped alignments was set to 2 000 bp.According to the alignment relationship observed in Figure 1C, these contigs perfectly matched our assembly, indicating an improved result.
To check the integrity and accuracy of the assembledchromosome-level genome, we conducted related analysis.All stLFR reads were mapped to the chromosome-level genome assembly by SOAP, BWA, and SAMtools to check coverage,single nucleotide polymorphisms (SNPs), and GC content (Li et al., 2009).The high mapping rate, high coverage, and concentrated distribution of GC content plot all indicated an accurate and precise genome.Related results are shown in Figure 1D.As an important evaluation index, BUSCO(Benchmarking Universal Single-Copy Orthologs) is widely used to quantitatively assess genome assembly and annotation completeness based on evolutionarily informed expectations of gene content.We chose the actinopterygii_odb9 dataset, which is a widely recognized dataset in teleost studies.The benchmark of our genome reached 93.8%, suggesting almost complete assembly.
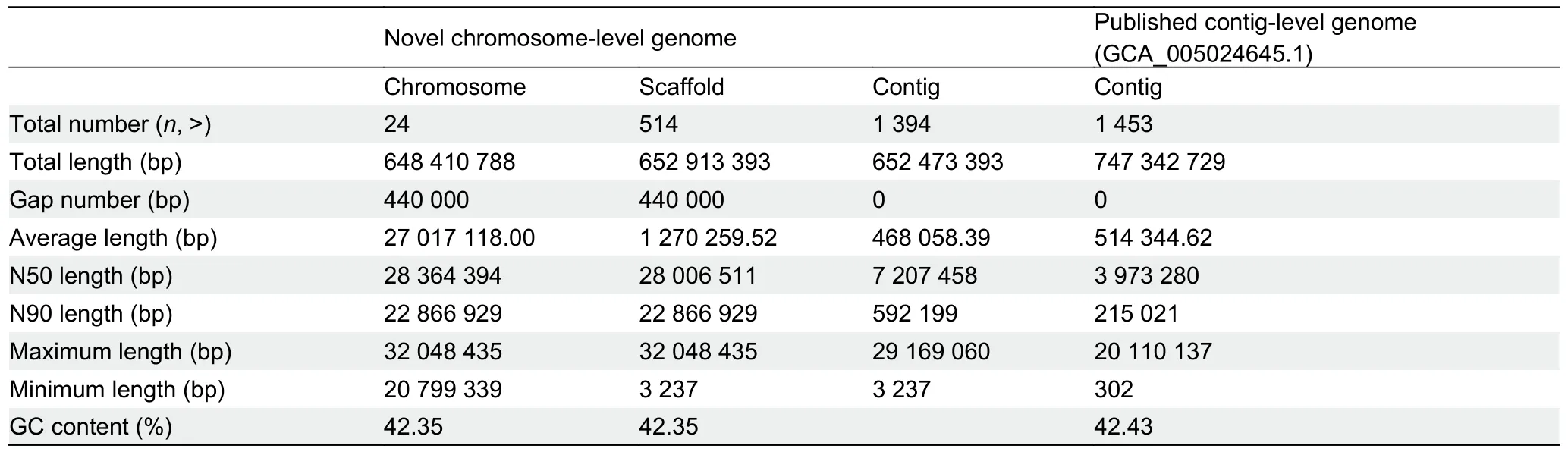
Table 1 Summary of information on genome assembly
After the genome assembly and evaluation pipeline, we carried out comprehensive annotation, including repeat sequences, gene structures, and gene functions.Genomic repetitive elements were identified with RepeatMasker(http://repeatmasker.org/RMDownload.html) and RepeatProteinMask using homology predictions based on RepBase (http://www.girinst.org/repbase).RepeatModeler(http://repeatmasker.org/RepeatModeler/), LTR_FINDER, and TRF tool were also used fordenovoprediction of repeat elements based on the features of the repeat sequences.The repeat libraries predicted bydenovoand homology methods were used to mask the repetitive regions of the genome via RepeatMasker prior to gene structure annotation.The repeat elements totaled 182 Mb, covering 27.99% of the genome, as predicted by TRF (2.20%), RepeatMasker (9.66%),RepeatProteinMask (3.34%), anddenovo(25.38%).
Genome structure and annotation analyses were performed using both homology anddenovomethods.BLAT was first employed to map the genome assembly to several highquality teleost protein assemblies, includingCynoglossus semilaevis,Daniorerio,Gadusmorhua,Gasterosteus aculeatus,Oreochromisniloticus,Oryziaslatipes,Seriola lalandi, andTakifugurubripes.The predicted gene structure models for these fish were defined by GeneWise to obtain a GFF file.To carry outdenovoprediction, we used Augustus,Genscan, and GlimmerHMM.All predicted gene models were combined by GLEAN to obtain the final genome structure.Combined results were then filtered to obtain more credible gene models.For example, if a predicted gene was only supported bydenovoevidence, then it had to be supported by all threedenovosoftware programs, or it was discarded.In total, 21 094 genes were predicted.We evaluated the exon number and length of different gene structures in the annotation data.Comparing our annotation to several reference species used in homology prediction, our results were relatively consistent (Figure 1E).
For functional annotation of the gene models predicted above, the Kyoto Encyclopedia of Genes and Genomes(KEGG), SwissProt, and TrEMBL (https://www.uniprot.org/statistics/TrEMBL) databases were employed using BLASTP with an E-value cutoff of 1E-5 (UniProt Consortium, 2018).InterPro was used to predict gene function at the domain level,and Gene Ontology (GO) annotation was also performed.Overall, 21 045 genes were functionally annotated, accounting for 99.77% of all genes predicted.
We also performed phylogenetic analysis ofP.haematocheilawithC.semilaevis,D.rerio,Gadusmorhua,Oreochromisniloticus,Oryziaslatipes,S.lalandi,T.rubripes,Mugilcephalus(unpublished data), andLepisosteusoculatus.We obtained related protein sequences and aligned them using BLASTP.The alignment results were then clustered with TreeFam, which was used for grouping orthologous protein sequences.Clustering results are shown in Figure 1F.
Based on single-copy orthologs, we carried out phylogenetic analysis using gene coding sequence (CDS), protein sequences, and Fourfold Degenerate Synonymous Sites(4DTv).We selected the most reasonable evolutionary tree from the optional method, including maximum-likelihood,Bayes, and RAxML, by comparing the topological structure of our tree to published trees.The phylogenetic tree and orthologous gene sets identified in CDS sequence analysis were used for divergence time estimation with PAML MCMCTree (http://abacus.gene.ucl.ac.uk/software/paml.html).Several key node times used for correction were found at TimeTree (http://www.timetree.org/).Both the tree and species divergence time are shown in Figure 1G.Results indicated thatP.haematocheilaandM.cephalusare quite evolutionarily close and diverged ~32.8 million years ago (Mya) (95% fiducial range 27.8–41.6 Mya).
After the phylogenetic tree was established, we use Computational Analysis of gene Family Evolution (CAFE) to analyze gene family expansion or contraction among the species mentioned above (Han et al., 2013).A higher frequency of gene contraction than gene expansion has been observed in earlier research (Olson, 1999), thus significantly more gene copies in a family may indicate that the gene family is involved in a specific function.This may provide a hint for downstream research and may be the key to environmental suitability or biological characteristics.
Among the 425 expanded gene families inP.haematocheila, 158 were changed significantly (ViterbiP<=0.05) and involved 859 genes.We performed GO and KEGG enrichment analyses of these related genes to determine the possible function of the gene families(Supplementary Tables S1, S2).The GO and KEGG enrichment results are shown in Figures 1H and 1I.Clearly,immune and apoptosis-related gene families showed significant pathway expansion, including the NF-kappa B signaling pathway, intestinal immune network for IgA production, antigen processing and presentation, Ras signaling pathway, NOD-like receptor signaling pathway, RIGI-like receptor signaling pathway, and Toll, Toll-like and Imd signaling pathway.Enrichment of genes in the oxygen binding pathway may indicate strong environmental adaptability, as observed in mullet fisheries and aquaculture.
We investigated positively selected genes ofP.haematocheiladuring evolution.We selectedC.semilaevis,Oreochromisniloticus,Oryziaslatipes,S.lalandi,P.haematocheila, andM.cephalusfrom the species above to narrow the scope of analysis.CodeML in PAML was employed to analyze the codon alignment results of the chosen species using the branch site model.In total, 1 981 genes inP.haematocheilawere recognized as positively selected and showed variable functions.The DNA replication helicase/nuclease 2 gene was among these genes, indicating thatP.haematocheilamay effectively repair DNA as an adaptation to stressful living environments.This study should be beneficial for downstream functional analysis of fish.
DATA AVAILABILITY
The genome assembly was submitted to the China National Gene Bank Database (CNGBdb: CNP0001604), National Center for Biotechnology Information (NCBI: PRJNA771825),and National Genomics Data Center (GSA: PRJCA006896).The annotation is available upon request.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
B.Z.and J.L.designed and supervised the study.H.B.G.and N.Z.performed computational analysis of stLFR, Hi-C,genome annotation, chromosome synteny analysis, and phylogenetic research.N.Z.and B.Z.wrote the manuscript.C.H.Z.and Q.X.D.edited the manuscript.All authors read and approved the final version of the manuscript.

- Zoological Research的其它文章
- Novel rhino-like SHJHhr mice with thyroid dysfunction
- A new species of Micryletta (Amphibia: Anura:Microhylidae) from the Langbian Plateau in southern Vietnam
- The high diversity of SARS-CoV-2-related coronaviruses in pangolins alerts potential ecological risks
- Site-specific and seasonal variation in habitat use of Eurasian otters (Lutra lutra) in western China:implications for conservation
- Understanding autism spectrum disorders with animal models: applications, insights, and perspectives
- Challenging Wallacean and Linnean shortfalls:Ectatosticta spiders (Araneae, Hypochilidae)from China